




















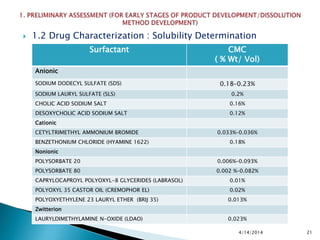






















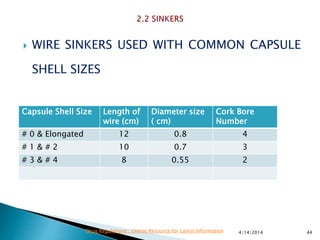




























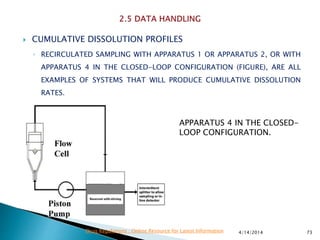





























































































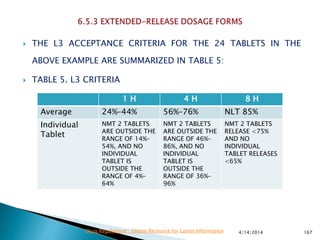


The presentation by 'Drug Regulations' outlines key aspects of pharmaceutical practices, including the importance of selecting suitable filter materials and methods for characterizing drug solubility and stability. It emphasizes the development and validation of dissolution methods for various dosage forms, highlighting the need for proper filtration techniques to ensure accurate results. Additional content includes guidance on selecting dissolution media and apparatus based on formulation type and solubility characteristics.






































![Analytical method validation raaj gprac [compatibility mode]](https://cdn.slidesharecdn.com/ss_thumbnails/analyticalmethodvalidationraajgpraccompatibilitymode-120830120141-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




































































