


























































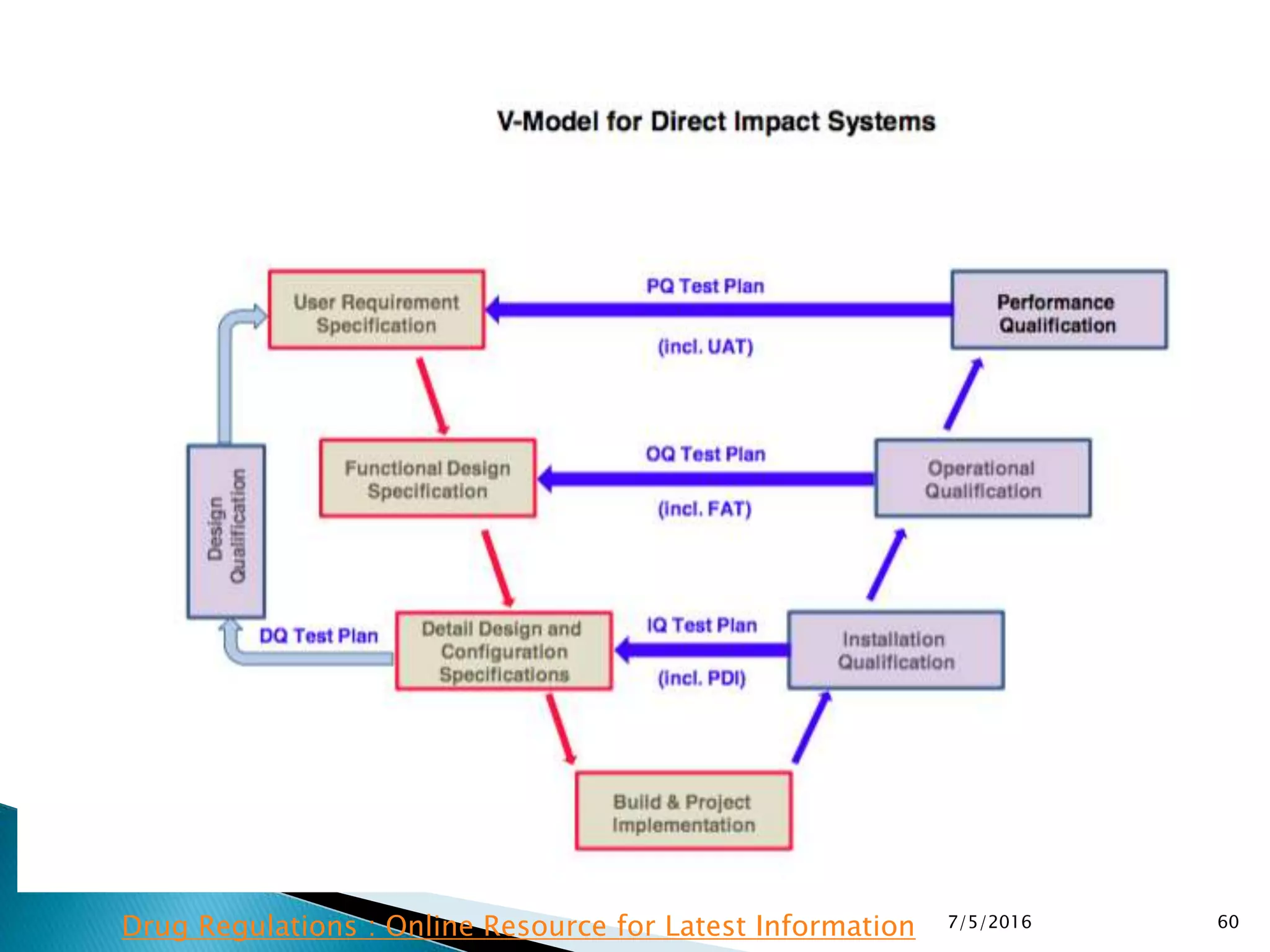
























This document provides guidance on validation and qualification principles from the World Health Organization (WHO). It discusses the need for validation and qualification activities to ensure product quality, safety, and efficacy throughout the product lifecycle. Key aspects covered include definitions of validation terms, approaches to validation planning, and documentation requirements such as a validation master plan and protocols.























































































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)