









































































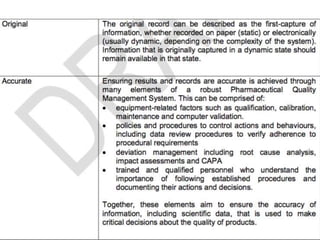






















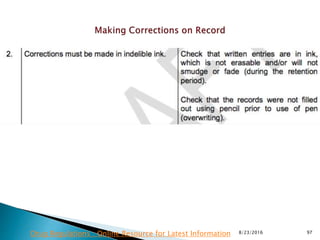
































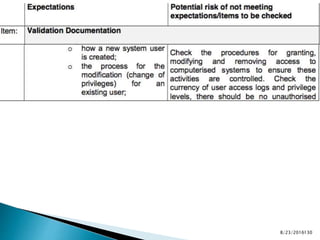



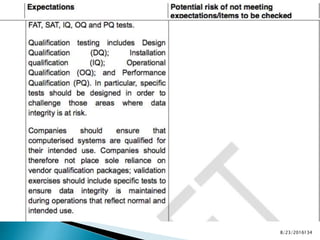




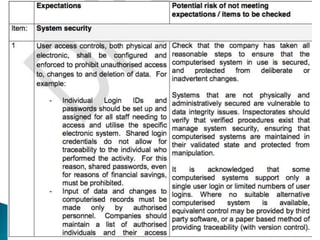





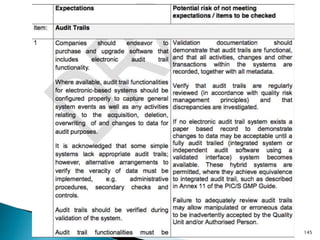






















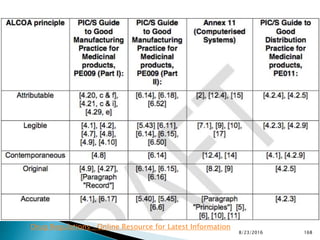

































This presentation by 'Drug Regulations' outlines principles and practices for ensuring data integrity in the pharmaceutical industry, particularly in contexts governed by Good Manufacturing Practices (GMP) and Good Distribution Practices (GDP). It emphasizes that data integrity is crucial for compliance and quality assurance, with responsibilities lying with manufacturers and distributors to manage their data governance systems effectively. The document examines how data governance systems should be integrated into organizational practices and stresses the importance of both automated and manual data processes.






















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)







![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)




![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)




