







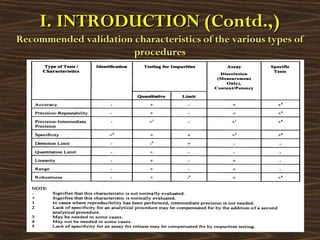


















![Different approaches suggested by ICH, USP & EP
Several approaches are given in the ICH guidelines for estimating LOD and LOQSeveral approaches are given in the ICH guidelines for estimating LOD and LOQ
Depending on the procedure : instrumental / non-instrumentalDepending on the procedure : instrumental / non-instrumental
ApproachApproach LODLOD LOQLOQ
Visual inspectionVisual inspection Minimum level detectableMinimum level detectable Minimum level quantifiableMinimum level quantifiable
Signal-to-Noise ratioSignal-to-Noise ratio 2:1 or 3:12:1 or 3:1 10:110:1
SD of response (SD of response (σσ) &) &
Slope (S)Slope (S)
[3:3 x[3:3 x σσ] / S] / S [10.0 x[10.0 x σσ] / S] / S
RSD CriteriaRSD Criteria Concentration at whichConcentration at which
RSD10 to 33.0%RSD10 to 33.0%
Concentration at which RSDConcentration at which RSD
≤≤10.0%10.0%
As per ICH and USP, other approaches suggested above are also
acceptable
2. LOD & LOQ (Contd.,)](https://image.slidesharecdn.com/ravisankar-130615222933-phpapp01/85/ANALYTICAL-METHOD-VALIDATION-BY-P-RAVISANKAR-27-320.jpg)

























This document discusses analytical method validation. It begins with an introduction that defines validation and discusses its importance and regulatory requirements. The document then covers specific validation parameters such as specificity, linearity, accuracy, precision, limit of detection, limit of quantification and more. For each parameter, the document provides definitions, procedures for evaluation, and acceptance criteria. It emphasizes that validation demonstrates a method is suitable for its intended purpose and supports the identity, quality, purity and potency of drug substances and products. The overall summary is that analytical method validation is critical to ensure quality and compliance in the pharmaceutical industry.











































































































