Downloaded 208 times
![Presented by:
K. ARSHAD AHMED KHAN M.Pharm, (Ph.D)
Department of Pharmaceutics,
Raghavendra Institute of Pharmaceutical Education and Research [RIPER]
Anantapur.](https://image.slidesharecdn.com/dissolutionchapter-4-180817070504/85/Dissolution-chapter-1-320.jpg)
![Presented by:
K. ARSHAD AHMED KHAN M.Pharm, (Ph.D)
Department of Pharmaceutics,
Raghavendra Institute of Pharmaceutical Education and Research [RIPER]
Anantapur.](https://image.slidesharecdn.com/dissolutionchapter-4-180817070504/75/Dissolution-chapter-1-2048.jpg)









































































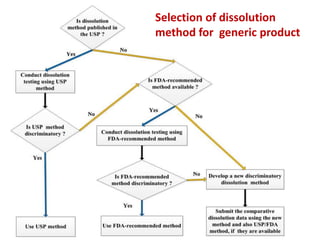











![1. Evaluation of the dissolution method
a) Drug substance solubility profile
b) Sink condition [are recommended, NOT necessary]
c) Justification and data to support selection of surfactant
[type, concentration]
d) Dissolution data in different pH media [pH 1.0, 4.5, 6.8]
without and with [if needed] surfactant
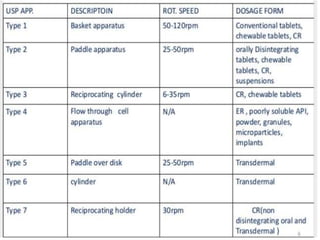
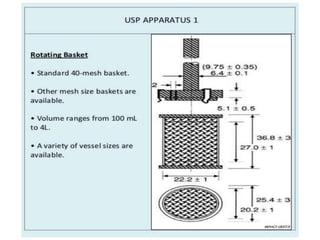
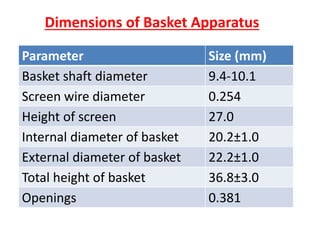
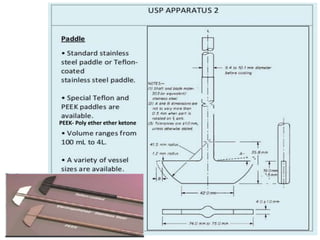
e) Selection of an appropriate apparatus/rotation speed.
f) Selection of in vitro dissolution/release medium/media
g) Selection of an appropriate analytical method](https://image.slidesharecdn.com/dissolutionchapter-4-180817070504/85/Dissolution-chapter-108-320.jpg)




This document focuses on dissolution testing in pharmaceutical development, detailing the equipment, calibration processes, and the importance of in-vitro dissolution tests for assessing drug quality and performance. It covers the historical evolution of dissolution methods, guidelines for developing and validating dissolution procedures, especially for generic drug products, and emphasizes the need for rigorous testing standards and methods. The document also outlines specific considerations for dissolution testing of various dosage forms, including recommendations for media, apparatus, and acceptance criteria.























































































