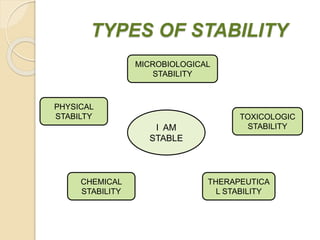
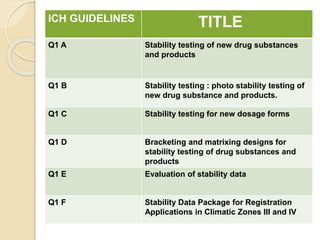
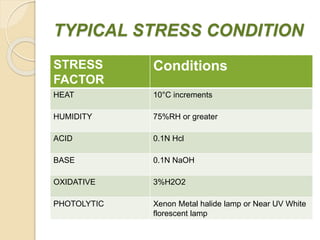
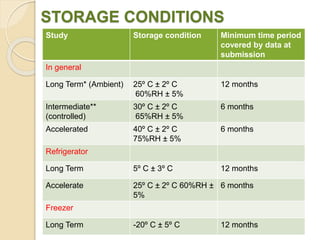
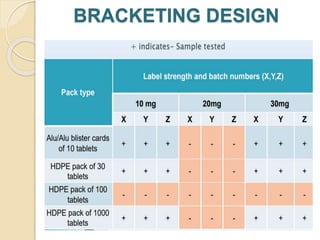
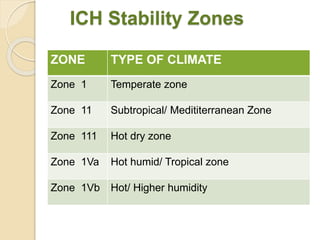
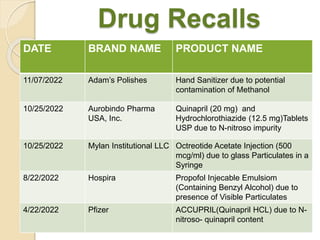
This document summarizes guidelines for stability testing of drugs and pharmaceutical products. It discusses how stability testing provides evidence on how a drug's quality varies over time under different environmental conditions, and enables establishing a shelf life. Key aspects covered include variables affecting stability, types of stability testing, ICH guidelines for stability testing terminology and procedures, storage conditions for long-term and accelerated testing, and evaluation of stability data. The goal of stability testing is to ensure drug quality is maintained throughout the proposed shelf life.




















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)







