






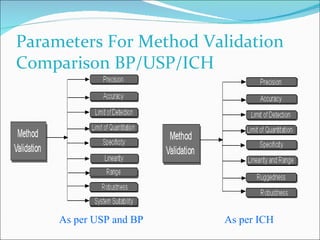

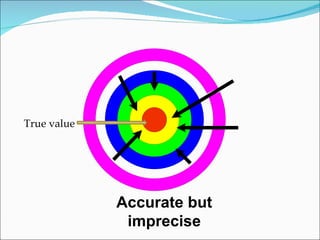


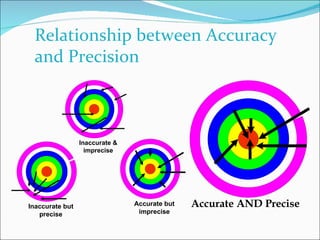
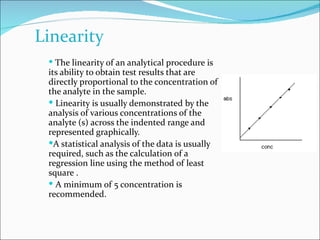








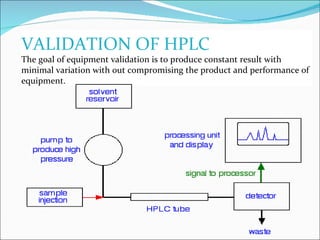




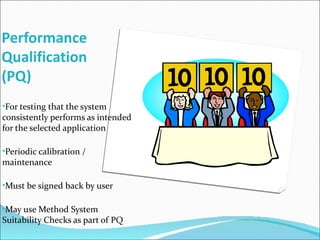




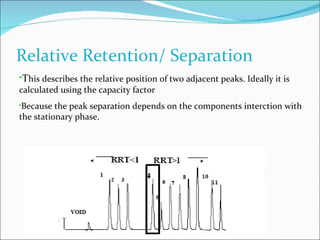

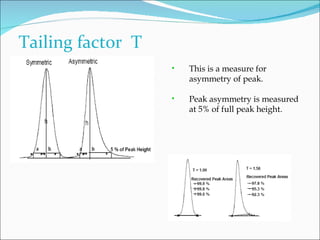






The document summarizes a seminar on analytical method validation and validation of HPLC. It discusses parameters for method validation according to USP, BP, and ICH guidelines such as accuracy, precision, linearity, range, specificity, detection limit, and quantitation limit. It also covers validation of typical HPLC systems through qualification, design, installation, operational, and performance qualification. Key parameters evaluated during HPLC method validation are discussed, including system suitability tests involving retention factor, relative retention, theoretical plates, resolution, and tailing factor.





























































































