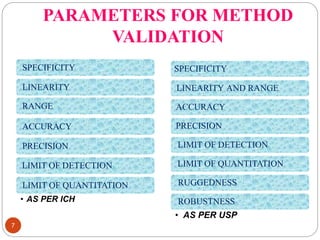
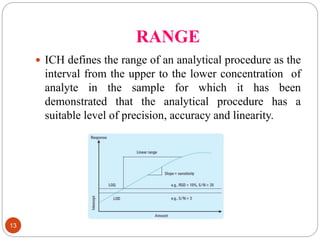
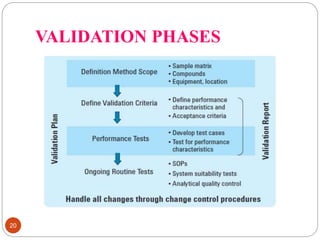
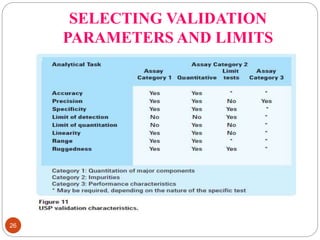
This document discusses analytical method validation. It defines analytical method validation as providing assurance that an analytical method can consistently and accurately determine the presence or quantity of attributes. The objectives of validation are to obtain consistent, reliable and accurate data. Key parameters that are assessed in validation include specificity, accuracy, precision, linearity, range, limits of detection and quantification, ruggedness and robustness. The validation process involves planning, testing method performance characteristics, selecting validation acceptance criteria, and documenting results in a validation report. Validation is important for analytical methods used in pharmaceutical analysis.