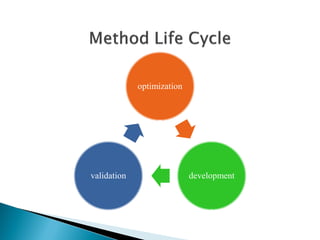
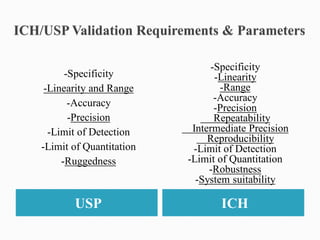
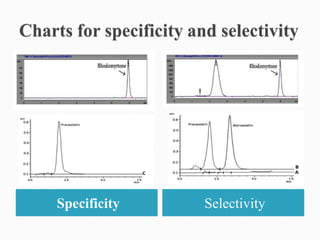
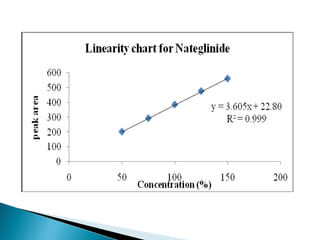
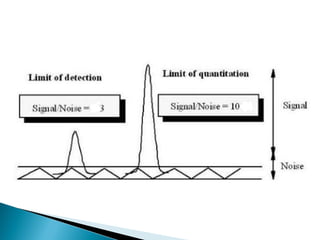
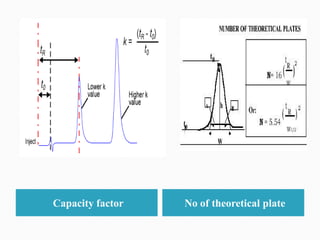
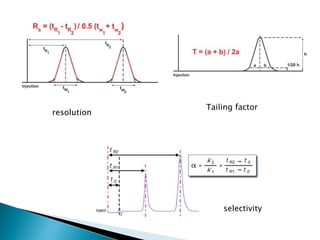
The document discusses validation of analytical procedures. Validation is required to confirm a procedure is suitable for its intended use. It identifies potential errors and determines if the method is acceptable. Key validation characteristics discussed include specificity, linearity, range, accuracy, precision, limit of detection, limit of quantitation, robustness, and system suitability. The document provides details on how to evaluate each characteristic.






































































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)
