Downloaded 443 times




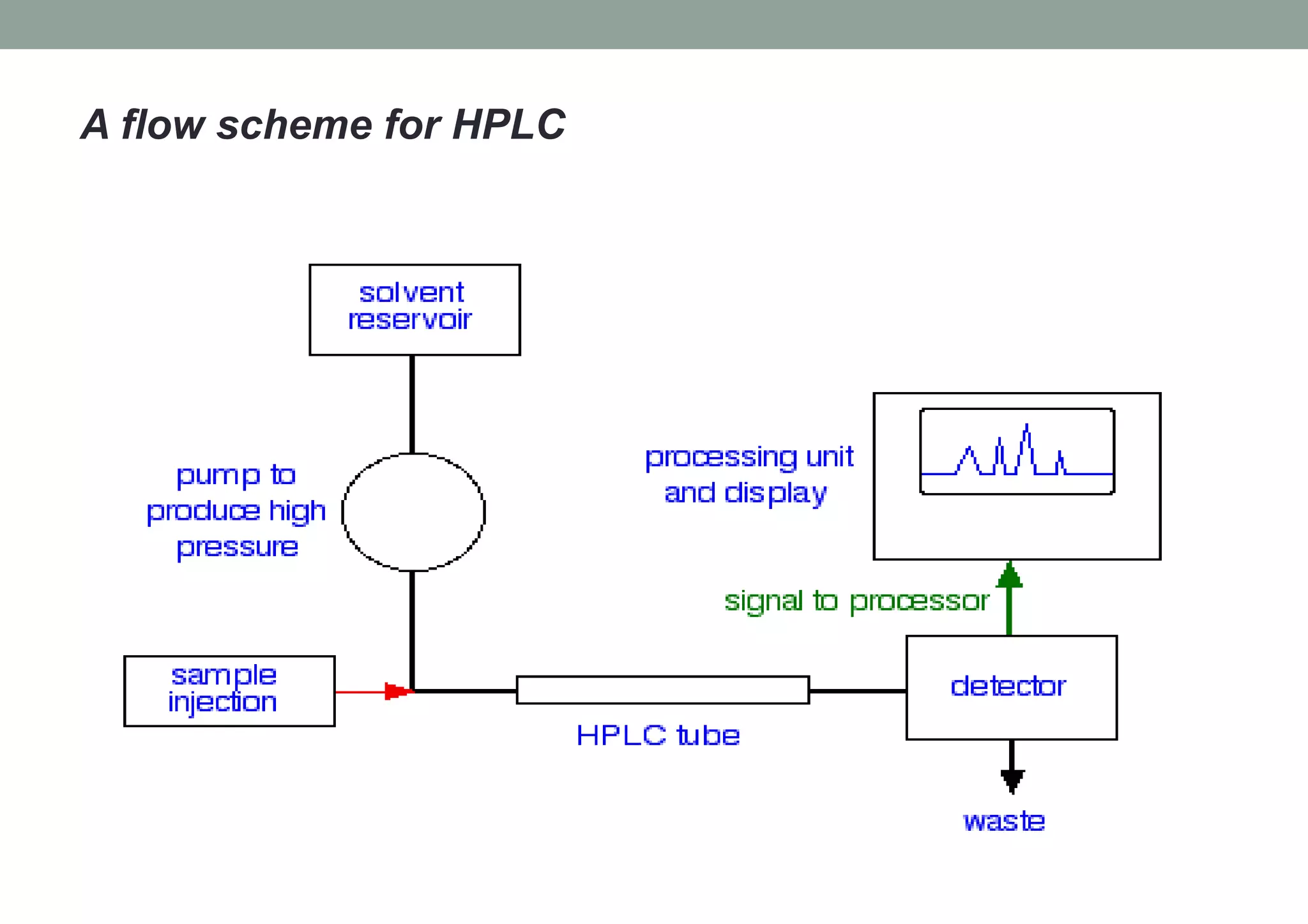









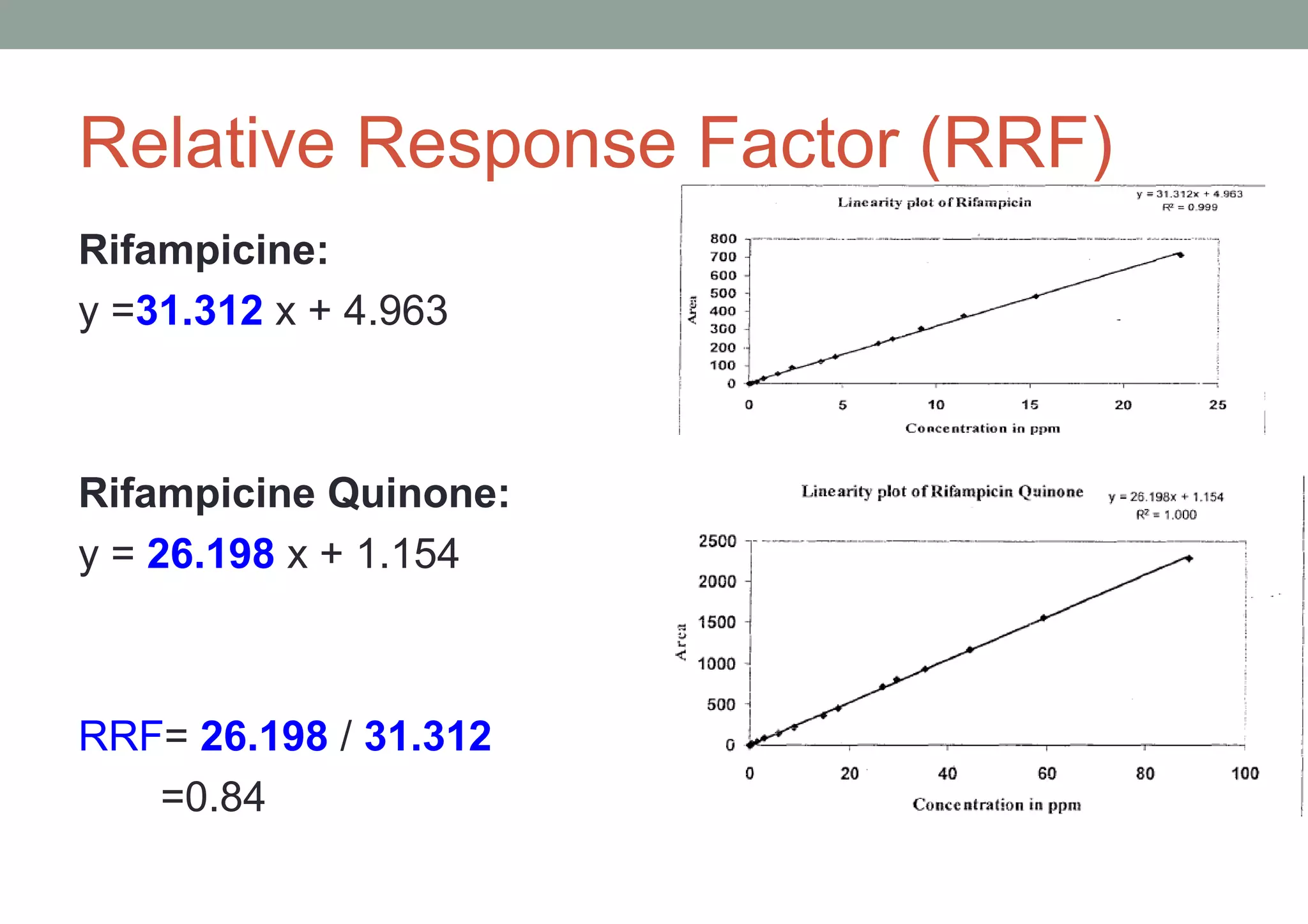




















This document provides an overview of HPLC methodology and validation requirements. It discusses the key components of an HPLC test procedure including system suitability testing, relative response factors, and the validation parameters of specificity, linearity, accuracy, precision, LOD/LOQ, and robustness. Validation requirements depend on whether the method is compendial or non-compendial, with full validation needed for non-compendial methods. System suitability criteria and validation acceptance limits are outlined for various analytical techniques like assay, impurities testing, and dissolution.



































































