Downloaded 571 times
















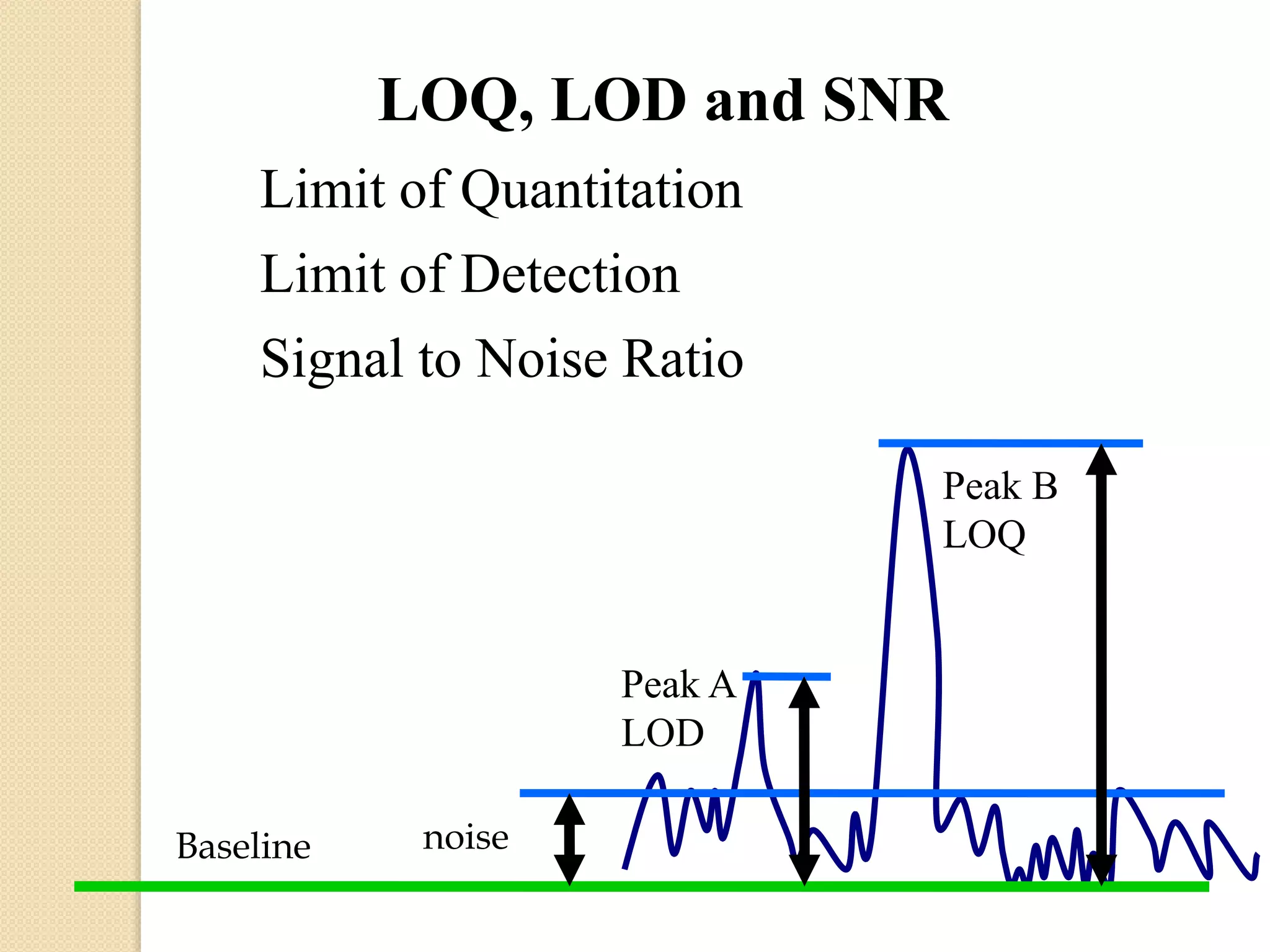




The document discusses analytical method validation. It defines validation as establishing evidence that a process will consistently produce a product meeting predetermined specifications. The objectives are to discuss aspects of validation including principles, approaches, and characteristics. Key steps in validation are establishing accuracy, precision, specificity, linearity, range, limits of detection and quantification, and robustness of analytical procedures used for identification, quantification of impurities and active ingredients.


















































































