Downloaded 463 times






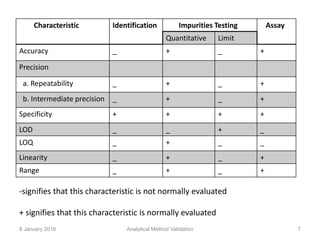

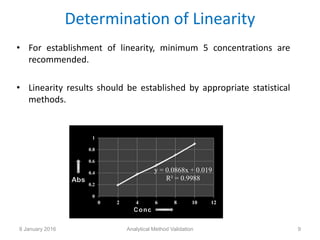






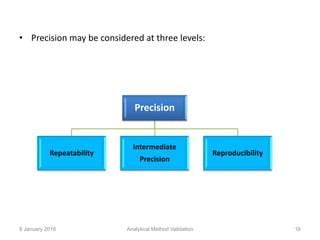



















Validation is defined as establishing documented evidence that a process will consistently produce results meeting pre-determined specifications. Key aspects of analytical method validation include accuracy, precision, specificity, limit of detection/quantitation, linearity, range, robustness, and system suitability. Validation demonstrates a method is suitable for its intended use and ensures consistent, reliable results are obtained in compliance with regulations.
















![Basics impurity profiling and degradent characterization[134]](https://cdn.slidesharecdn.com/ss_thumbnails/basicsimpurityprofilinganddegradentcharacterization134-191014164210-thumbnail.jpg?width=640&height=640&fit=bounds)
































































