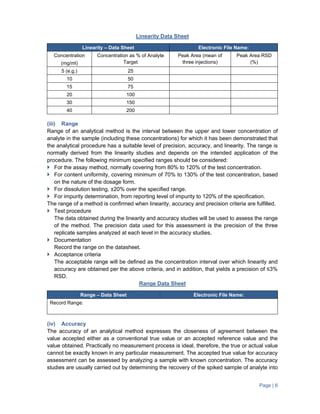
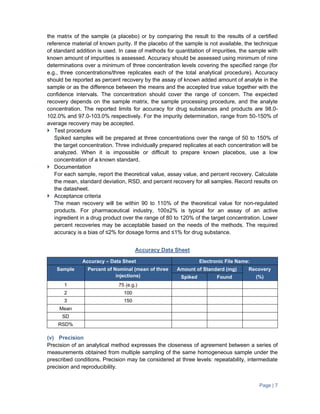
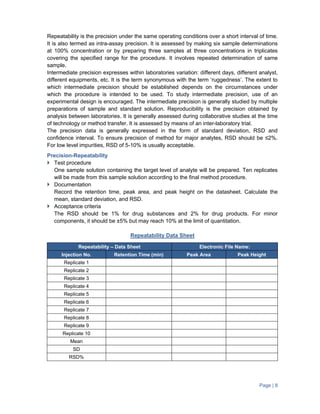
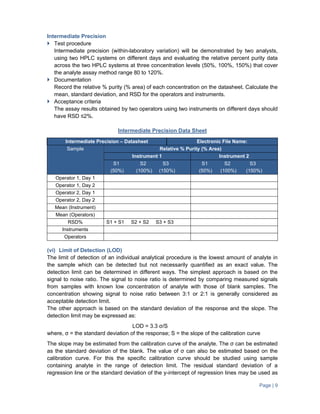
The document provides guidance on validation of analytical methods. It defines method validation as demonstrating that a method's performance characteristics meet requirements for intended use. All analytical methods for analyzing samples must be validated. The validation process involves developing a protocol, validating parameters like specificity, linearity, range, and accuracy, and documenting the results. Parameters to be validated depend on the type of test (e.g. assay, impurities identification). Acceptance criteria for characteristics like linearity, range, and accuracy are provided.