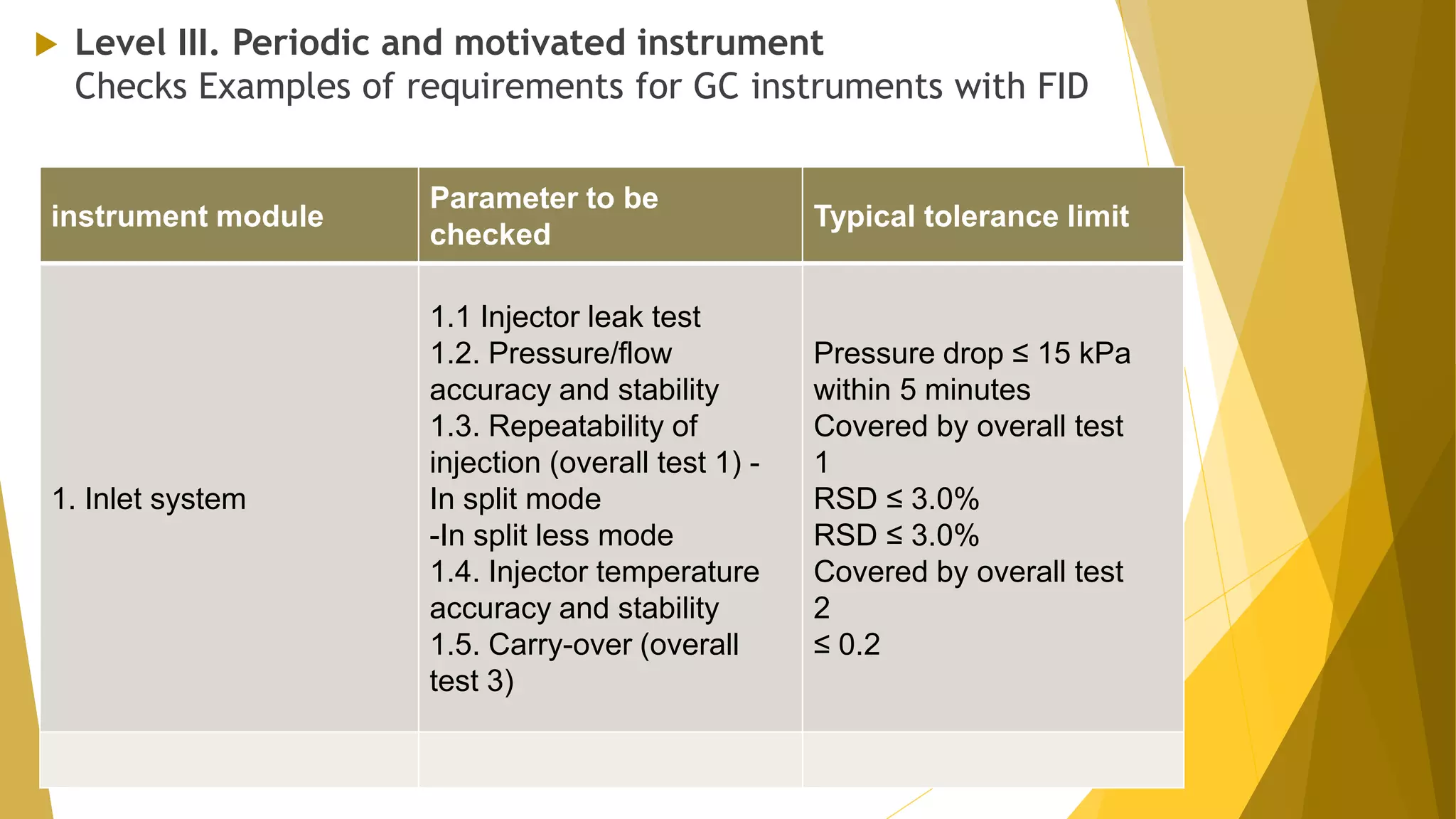
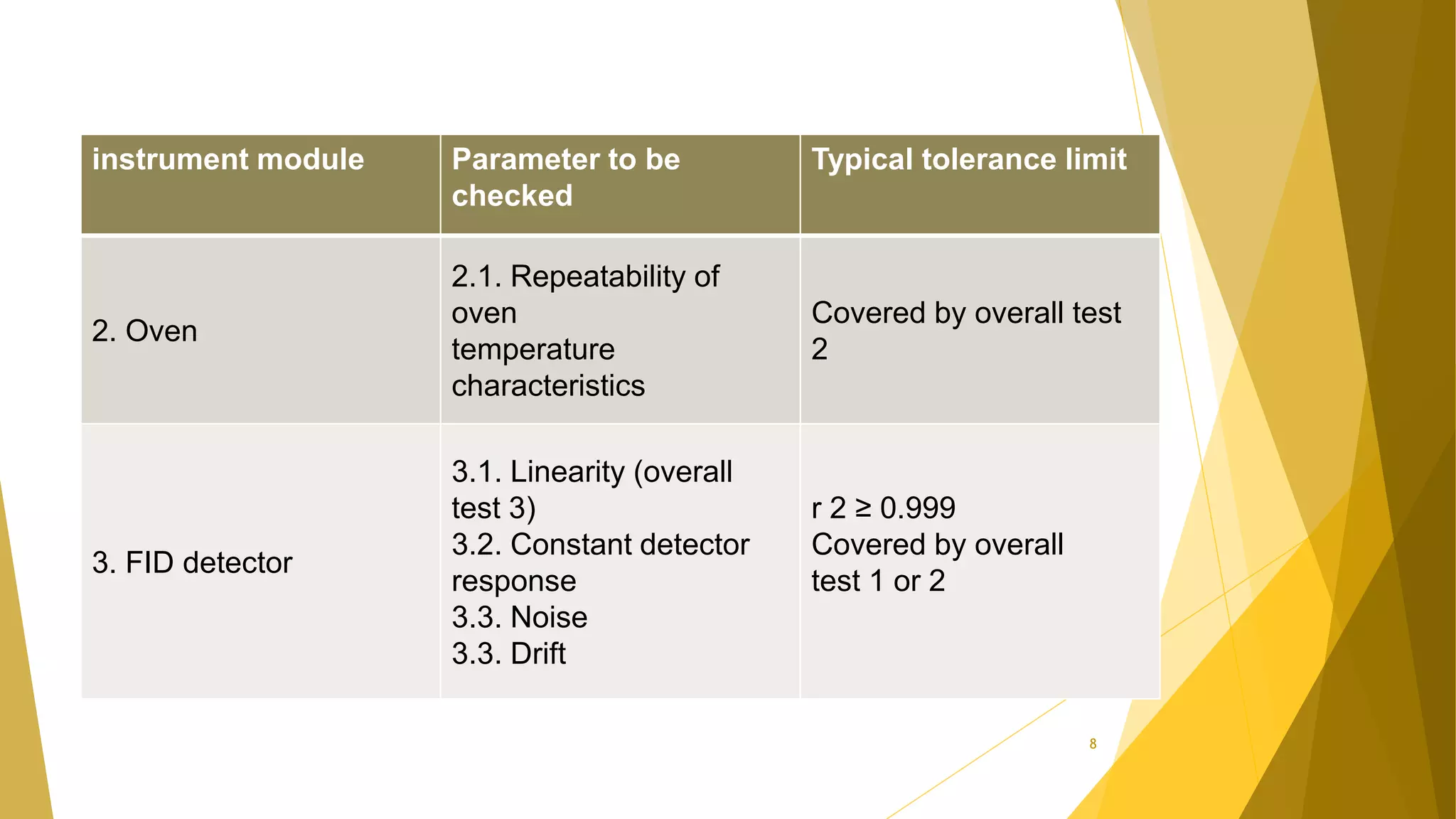
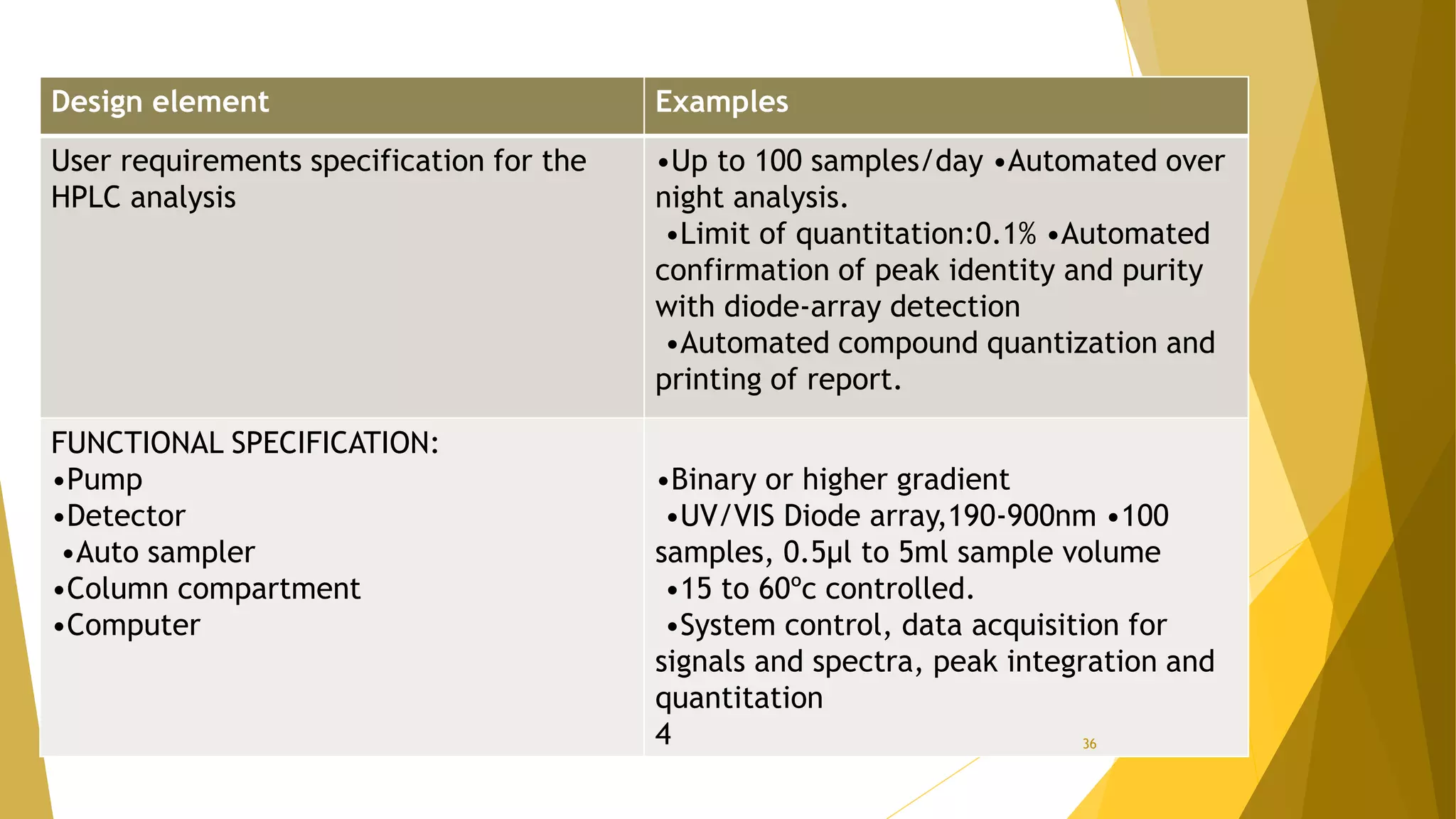
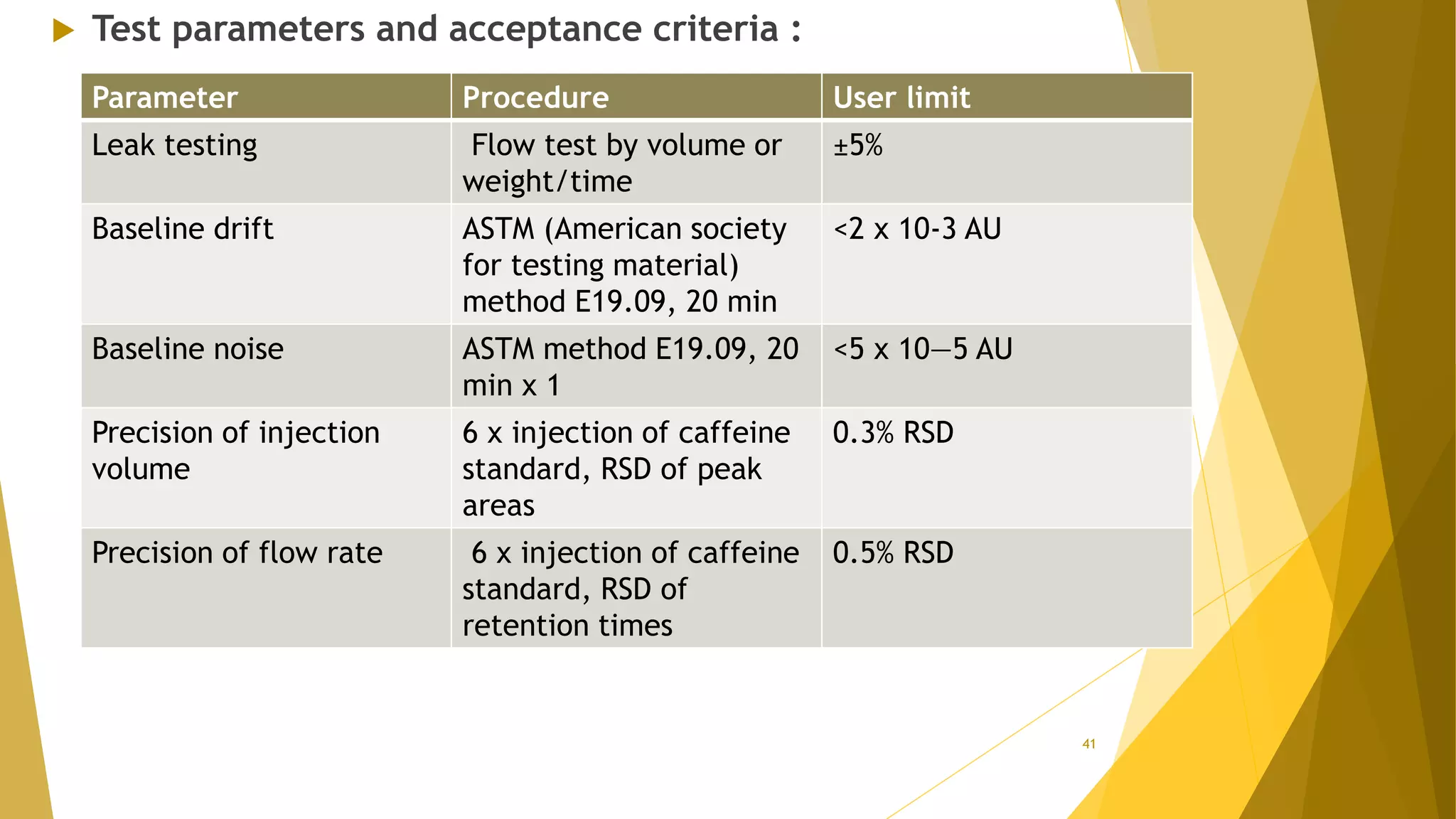
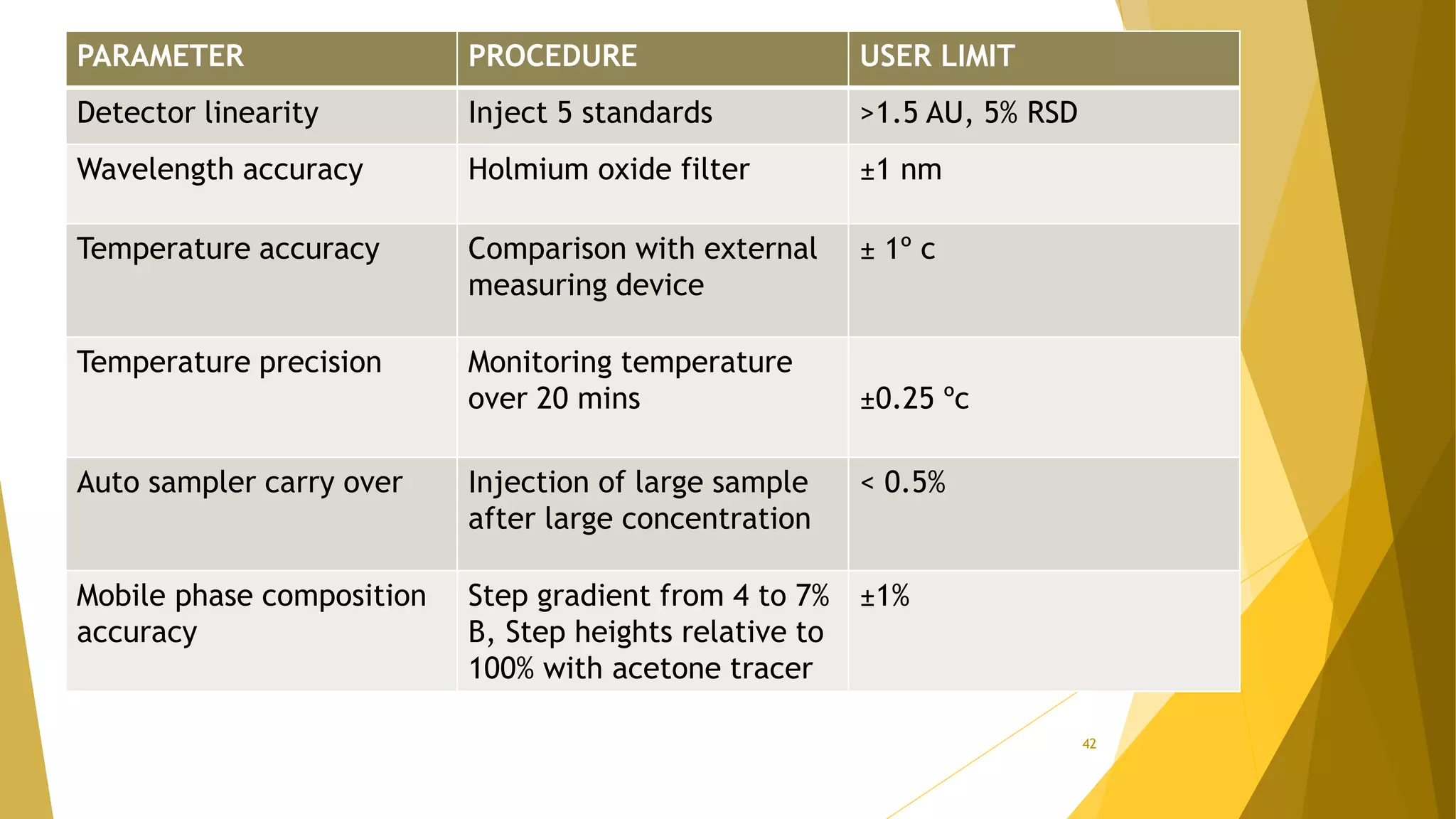
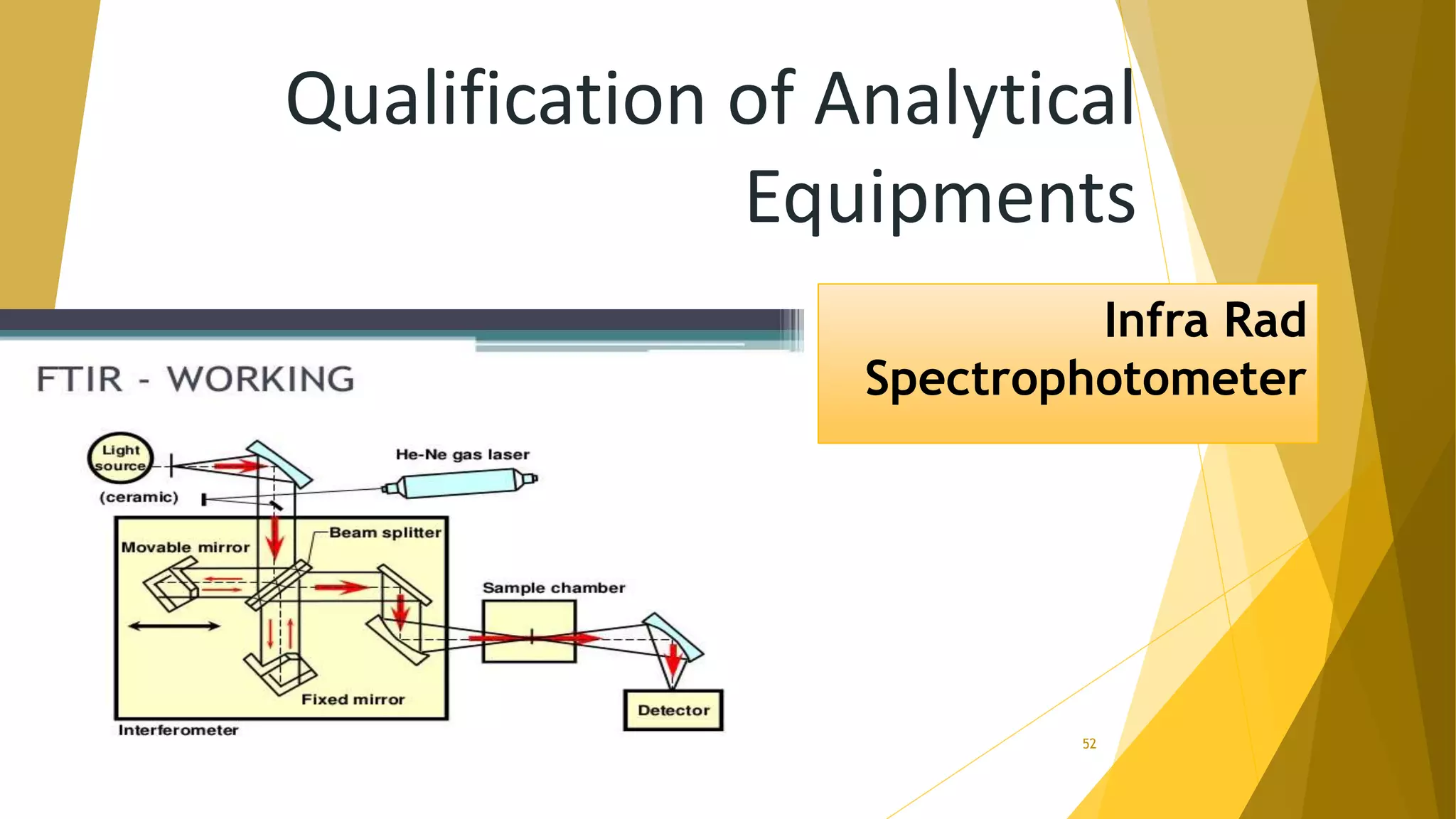
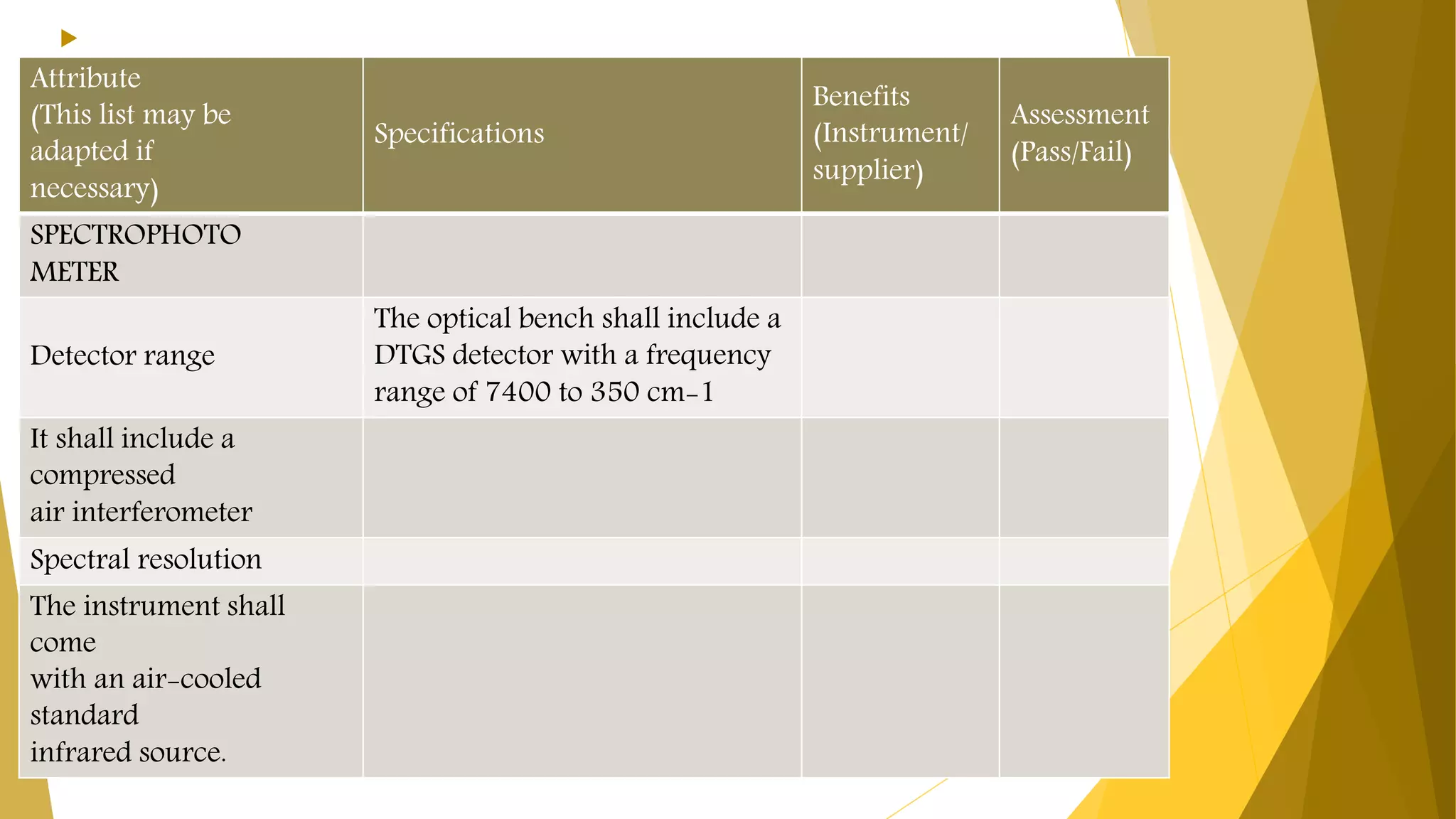
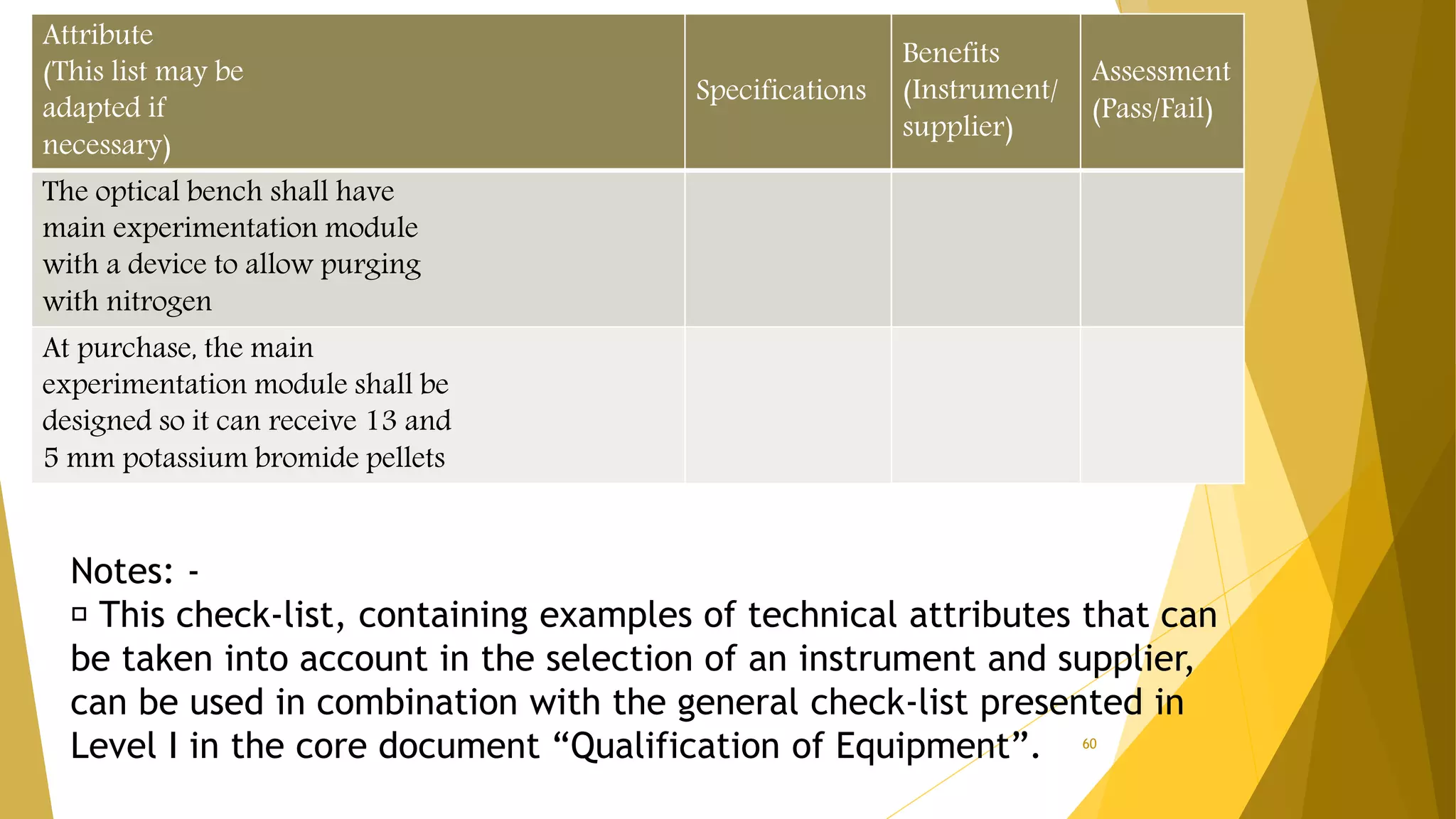
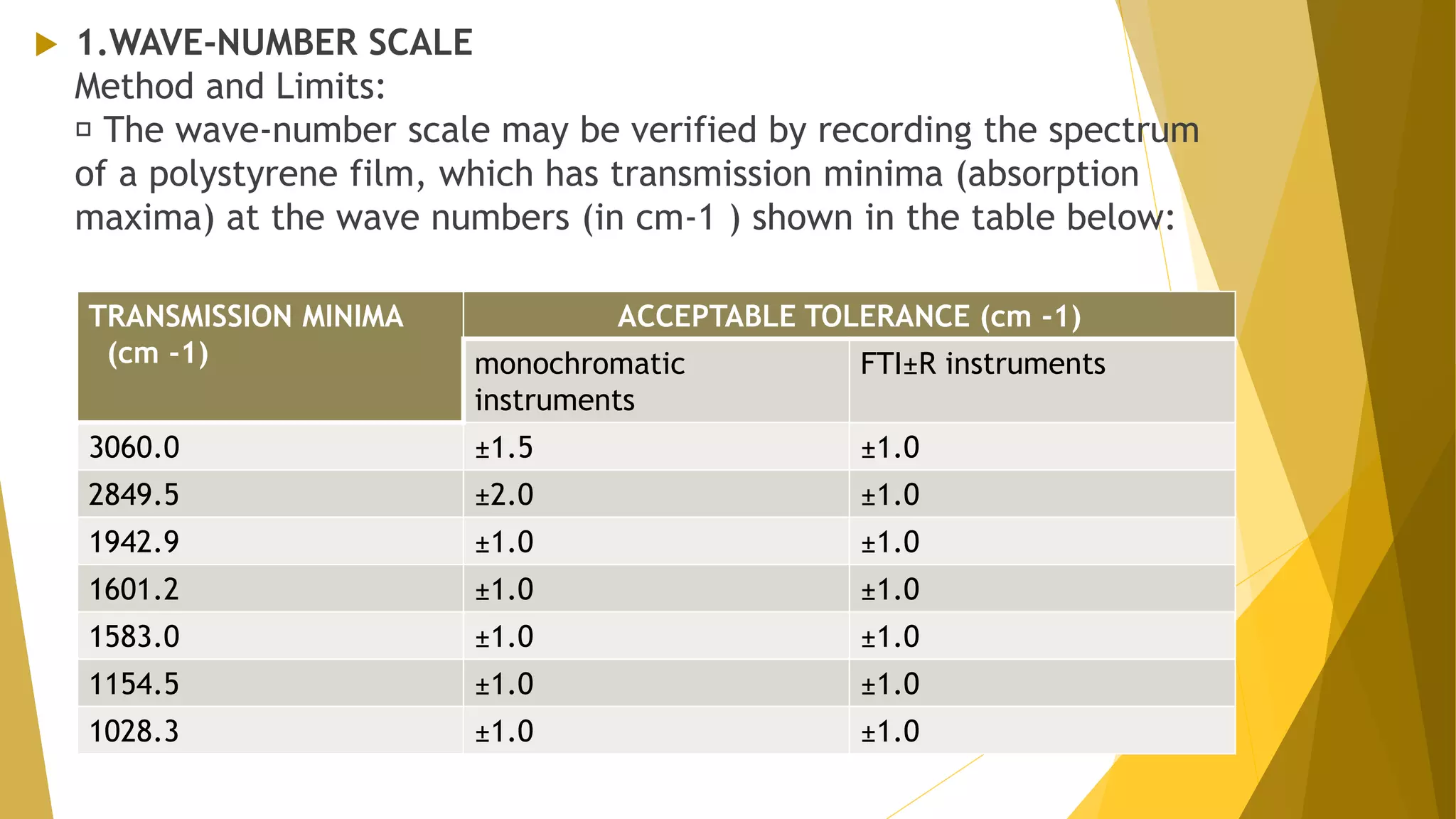
The document discusses the qualification of analytical equipment, specifically gas chromatography. It describes the four parts of qualification: design qualification, installation qualification, operational qualification, and performance qualification. It then provides examples of tests and acceptance criteria for various parameters of a gas chromatography system, including the inlet system, oven, and flame ionization detector. Tests include leak tests, repeatability, linearity, noise, drift and more. The goal is to verify that the GC system is installed and functioning properly.