Downloaded 138 times

















![ANALYTICAL METHOD VALIDATION
• What Is An ATP?
• Analytical Target Profile is a part of QbD which is a predefined,
documented statement of the requirements for the quality of the
reportable value, which the analytical procedure must generate in
order for the procedure to be considered fit for purpose Using
Current Industry Acceptance Criteria. The procedure must be able
to accurately quantify [drug] in the [FPP] in the presence of
[Impurity/degradation product] with the following requirements for
the reportable values: Accuracy = 100.0% ± 3.0% and precision ≤
2.0% relative standard deviation (RSD).
ANALYTICAL METHOD DEVELOPMENT
19](https://image.slidesharecdn.com/analyticalmethodvalidation-171230043307/85/Analytical-method-Content-Development-validation-Transfer-Life-Cycle-Management-19-320.jpg)

































































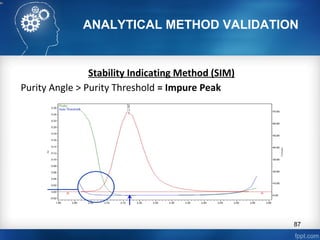
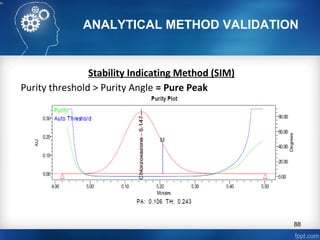
















This document discusses analytical method validation and provides guidelines on developing and validating analytical methods according to regulatory standards. It outlines the key components that should be included in an analytical method as well as considerations for method development such as selecting stationary and mobile phases, operating parameters, and evaluating method performance characteristics during development. The document also discusses best practices for transferring validated analytical methods between laboratories.



































































