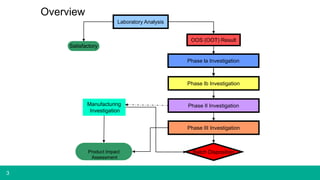
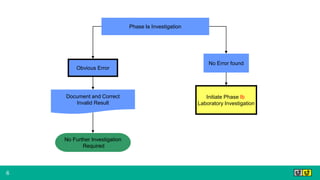
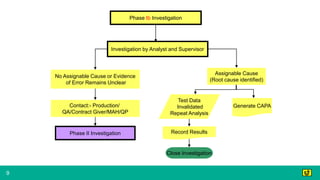
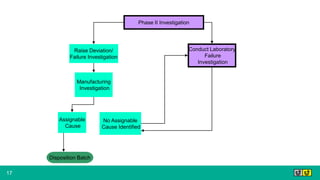
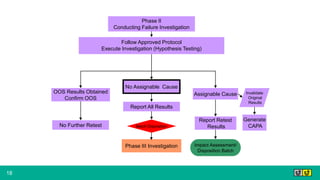
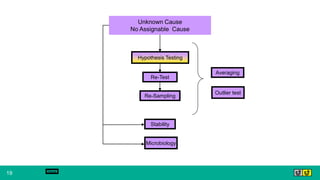
This document provides guidance on investigating out of specification or out of trend results from laboratory analyses. It describes a multi-step investigation process including: Phase Ia investigations to identify obvious errors; Phase Ib investigations conducted by an analyst and supervisor using a checklist to identify potential causes; Phase II investigations if no cause is found that may involve additional parties; and Phase III investigations and batch disposition if needed. Key terms related to investigations are also defined, such as out of specification, out of trend, assignable cause, and invalidated test.