Downloaded 21 times


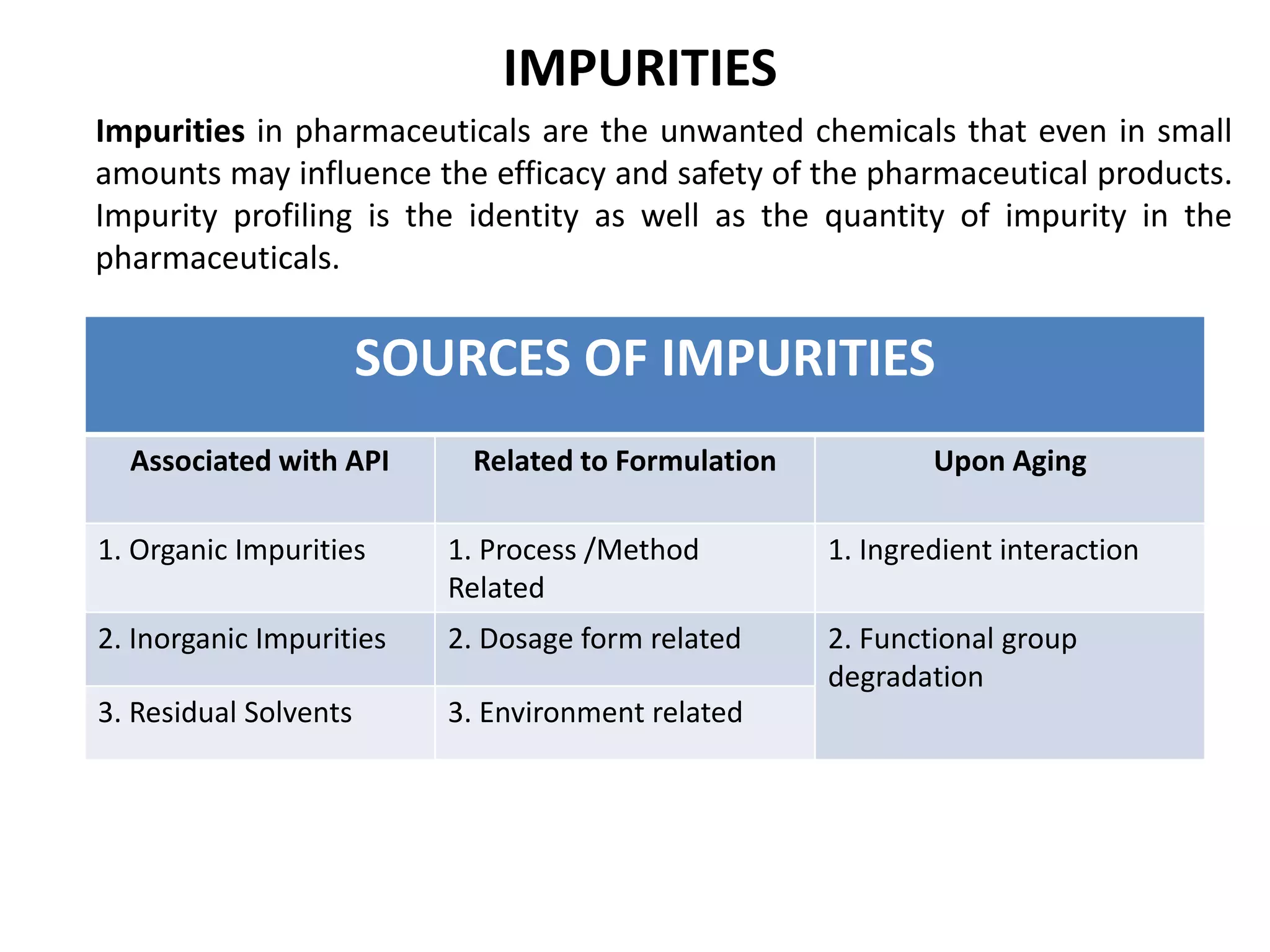



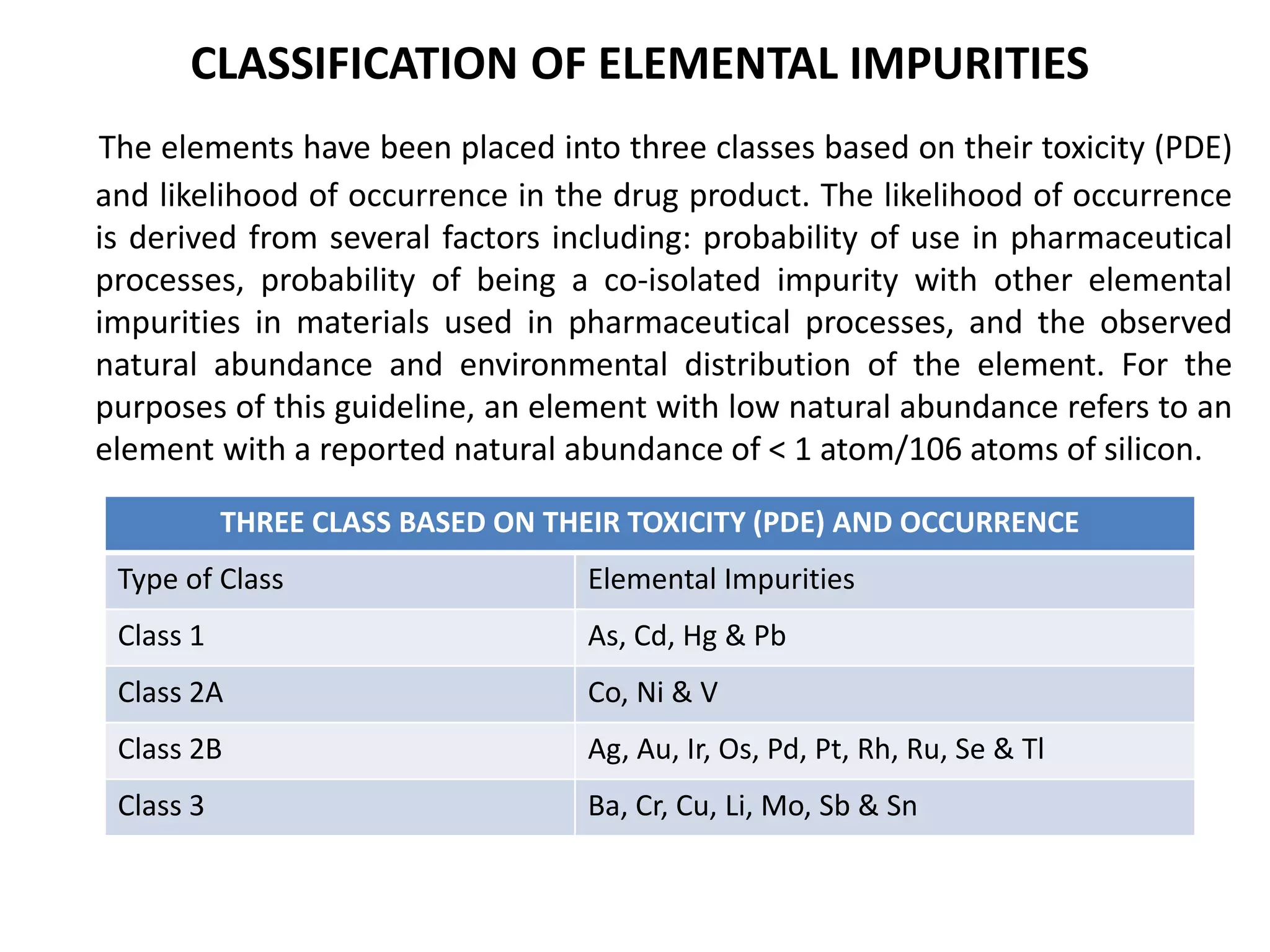



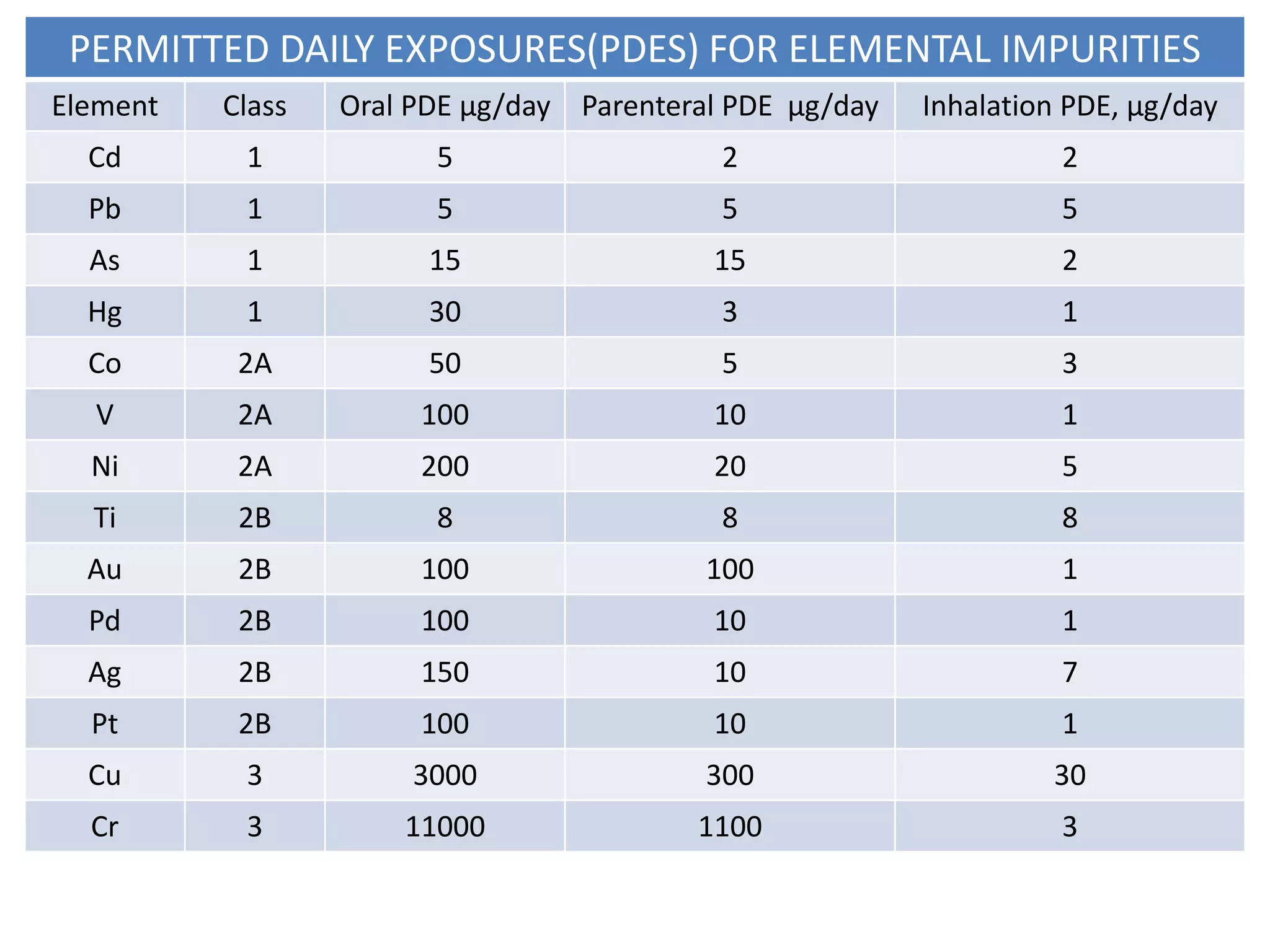

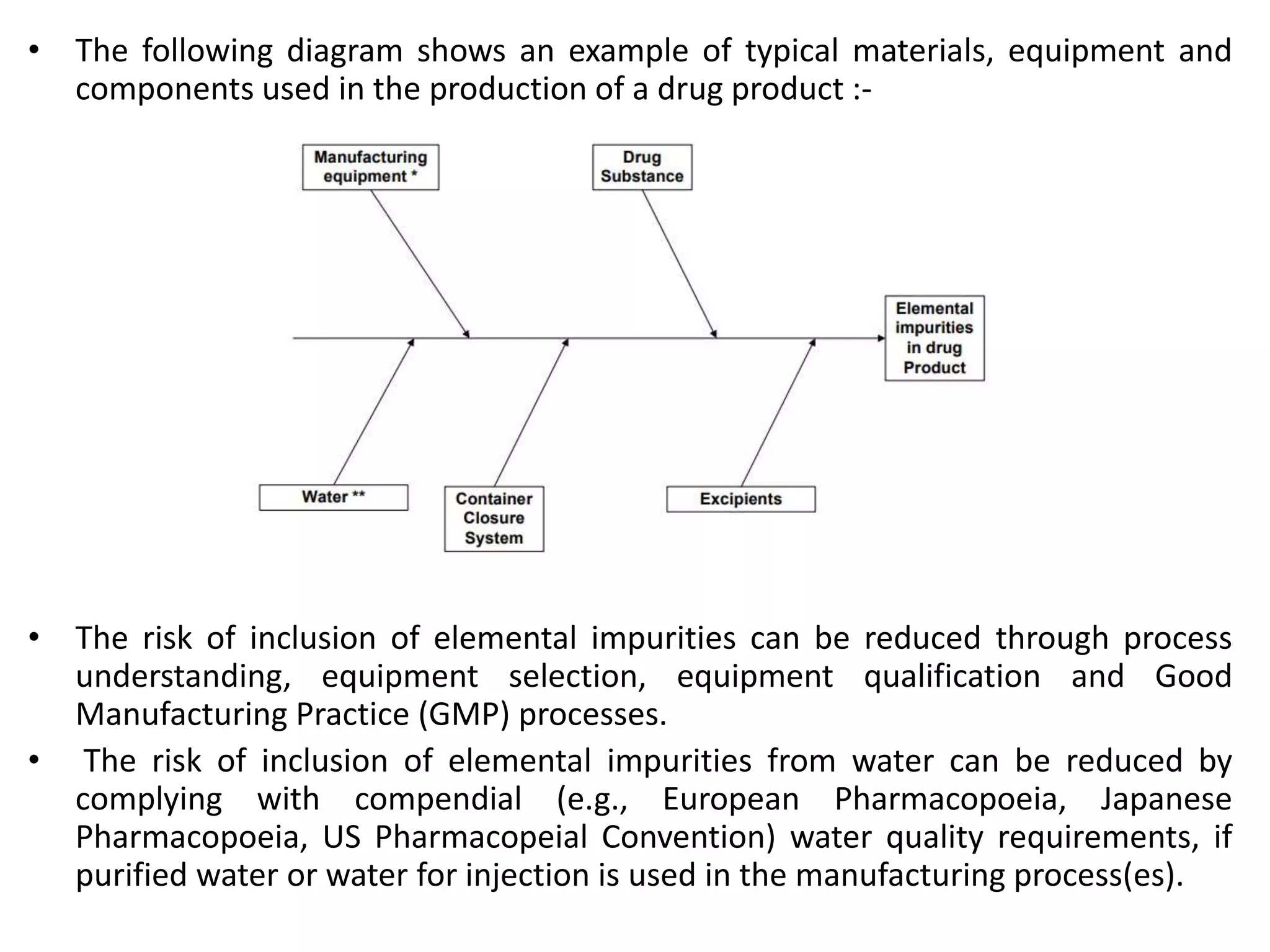








This document provides an overview of elemental impurities in pharmaceutical products. It discusses the ICH Q3D guideline for elemental impurities, including the classification of impurities into three classes based on toxicity and likelihood of occurrence. Potential sources of elemental impurities are identified, such as residual catalysts, excipients, manufacturing equipment, and container closure systems. The document also outlines the identification, control, and risk assessment of elemental impurities.



































































