Downloaded 2,820 times


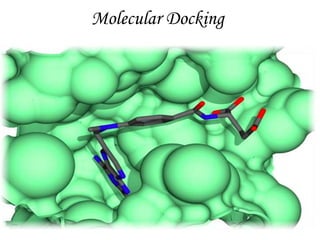




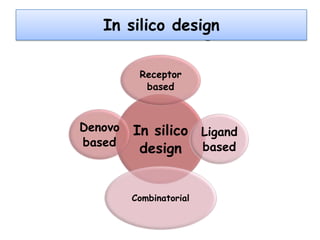
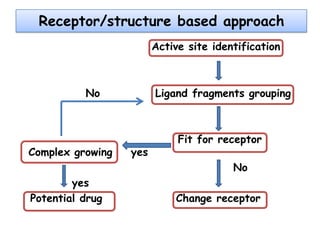
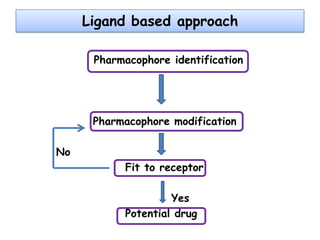




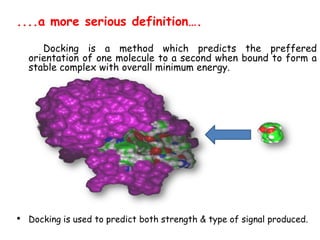







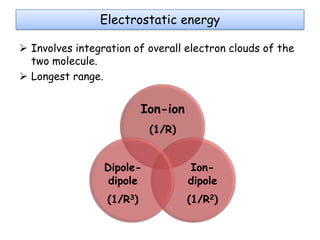



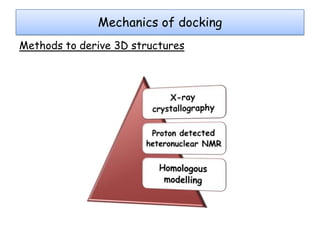
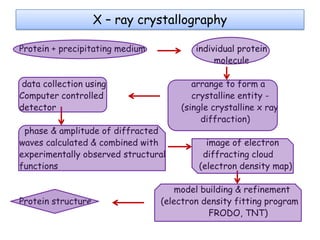
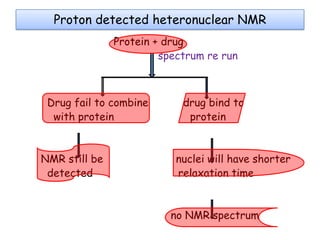

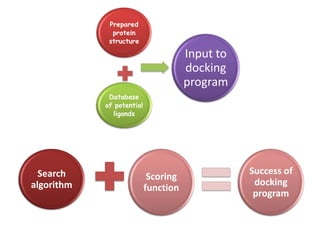

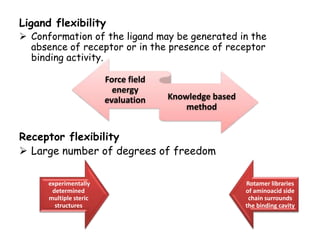
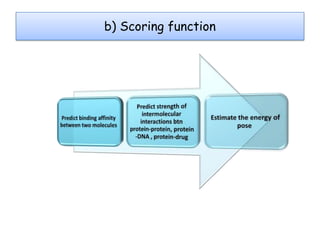
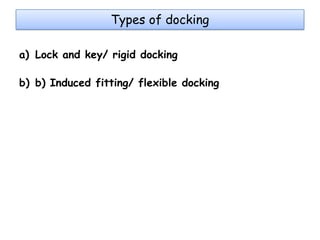

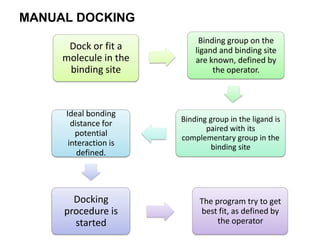
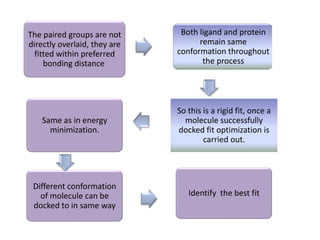



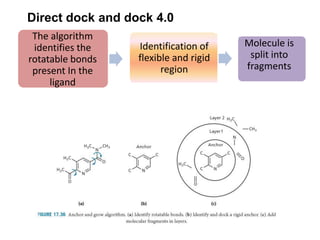
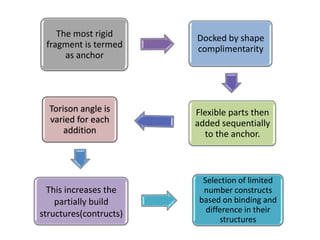


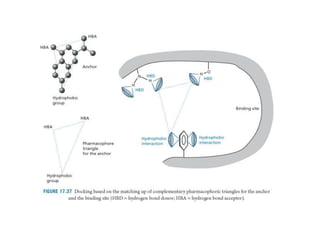

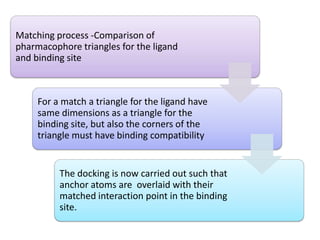
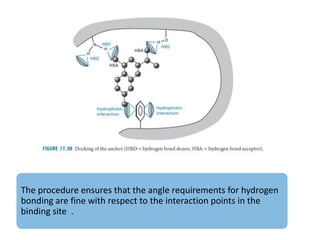

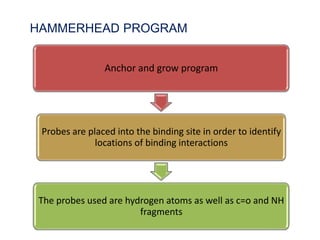


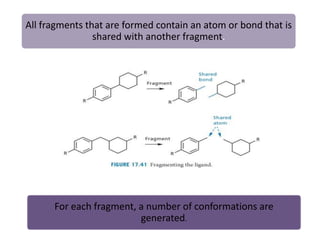
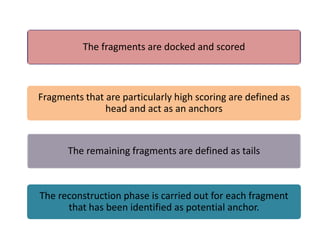
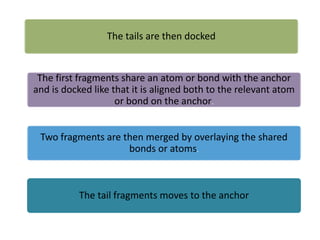
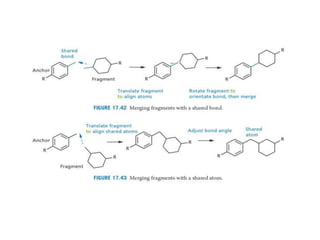












Molecular docking is a computer modeling technique used to predict the preferred orientation of one molecule to another when bound to form a stable complex. It involves fitting potential drug molecules into the active site of a protein receptor in order to identify which molecules may bind strongly. There are different approaches to molecular docking including rigid docking which treats molecules as rigid bodies, and flexible docking which accounts for conformational changes in ligands. The goal of docking is to find binding orientations that minimize the total energy of the system and maximize intermolecular interactions in order to predict effective drug candidates.
































































































