Downloaded 1,229 times


































![References
• Foye’s principles of medicinal chemistry-sixth edition,
published by lippincott williams and wilkins, page num-69-70
• Sanmati K jain and avantika agarwal- “ De novo drug design:an
overview”. Indian journal of pharmaceutical
sciences.,2004,66[6]:721-728
• http://www.bama.ua.edu/~chem/seminars/student_seminars
/spring06/papers-s06/luthra-sem.pdf
• http://journal.chemistrycentral.com/content/2/S1/
• http://en.wikipedia.org/wiki/HIV-1_protease
• Schneider G.; Fechner U.; “Computer-based de novo design of
drug-like molecules.” Nat Rev Drug Discov 2005 Aug; 4(8), 649-
63. Review](https://image.slidesharecdn.com/denovoppt-150424212022-conversion-gate02/75/Denovo-Drug-Design-37-2048.jpg)


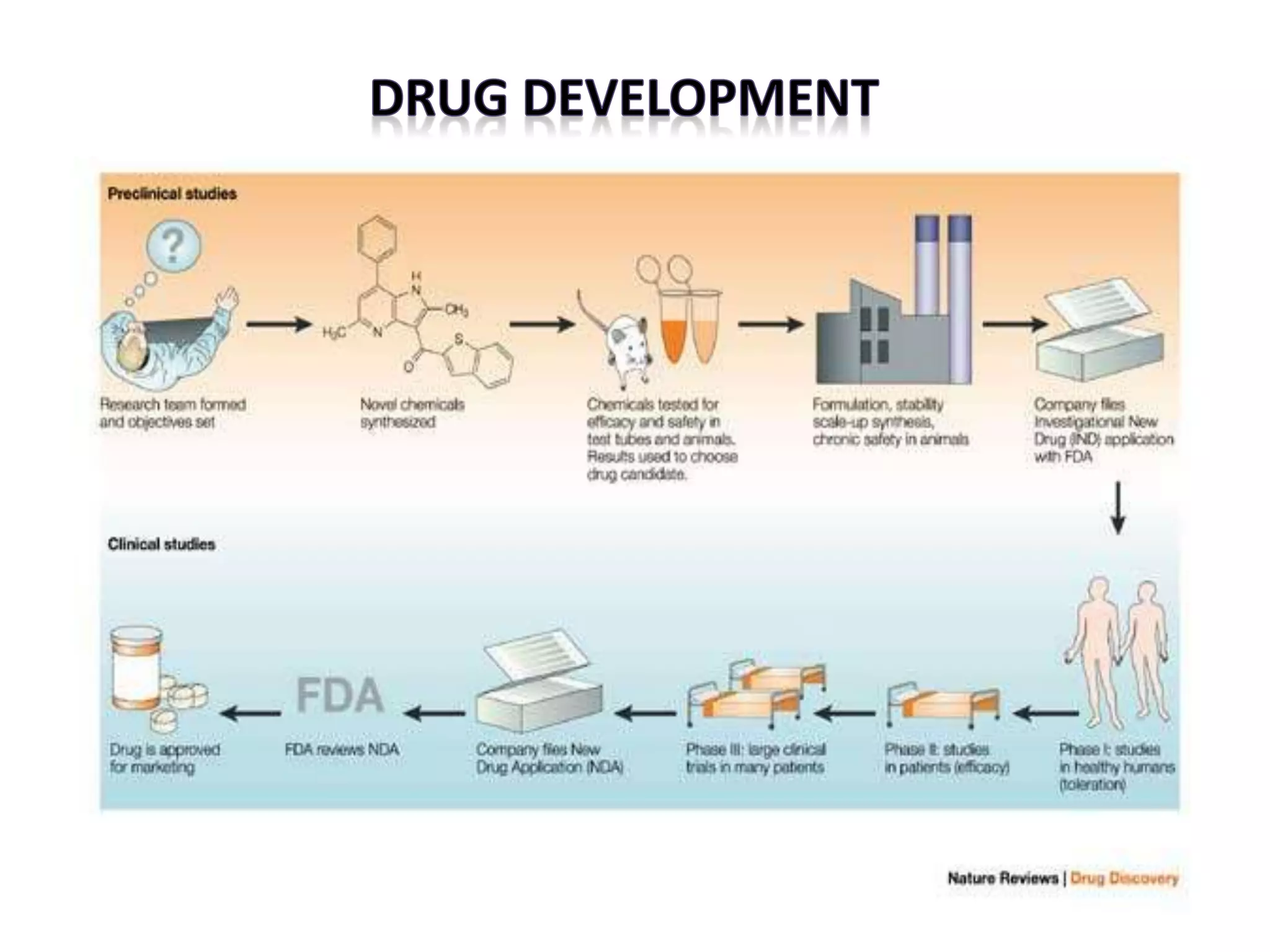
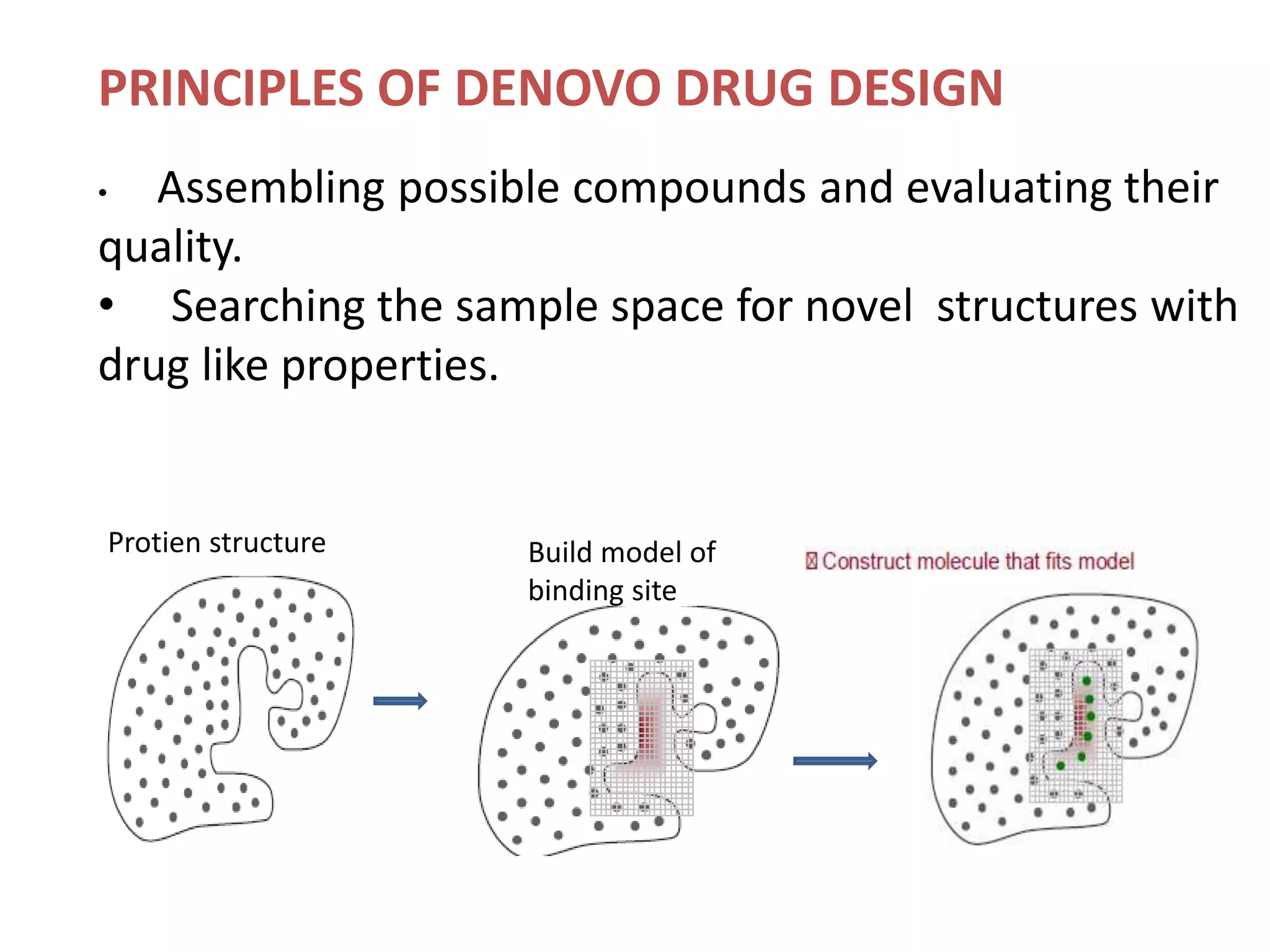
1) De novo drug design involves generating new drug molecules from scratch based on the 3D structure of the target receptor. 2) It uses molecular modeling tools to modify lead compounds to better interact with the receptor's binding site. 3) The process involves defining interaction sites on the receptor, generating potential drug molecules, scoring them based on their fit with the receptor, and using search algorithms to refine candidates.























































































