Download as PDF, PPTX



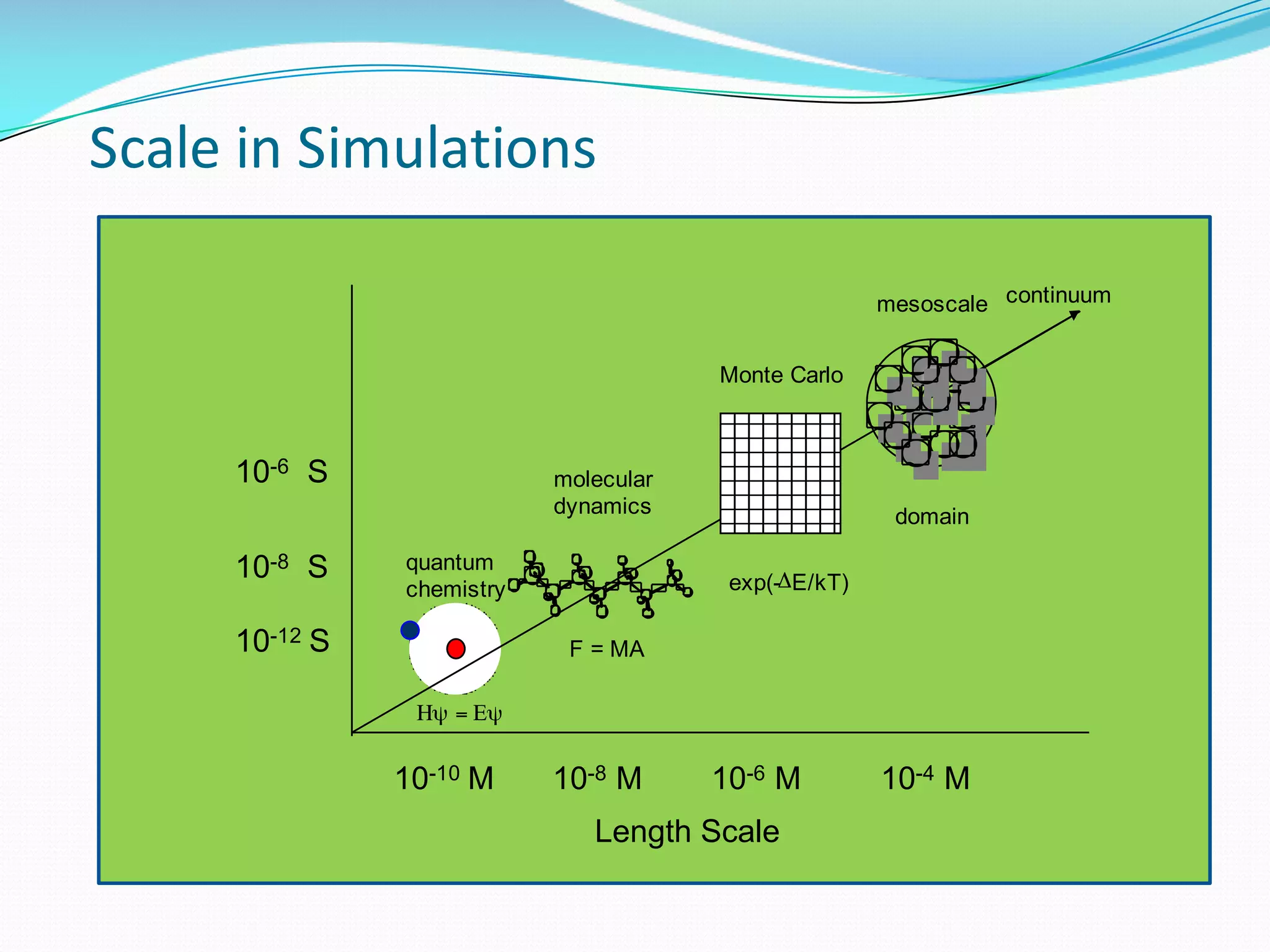


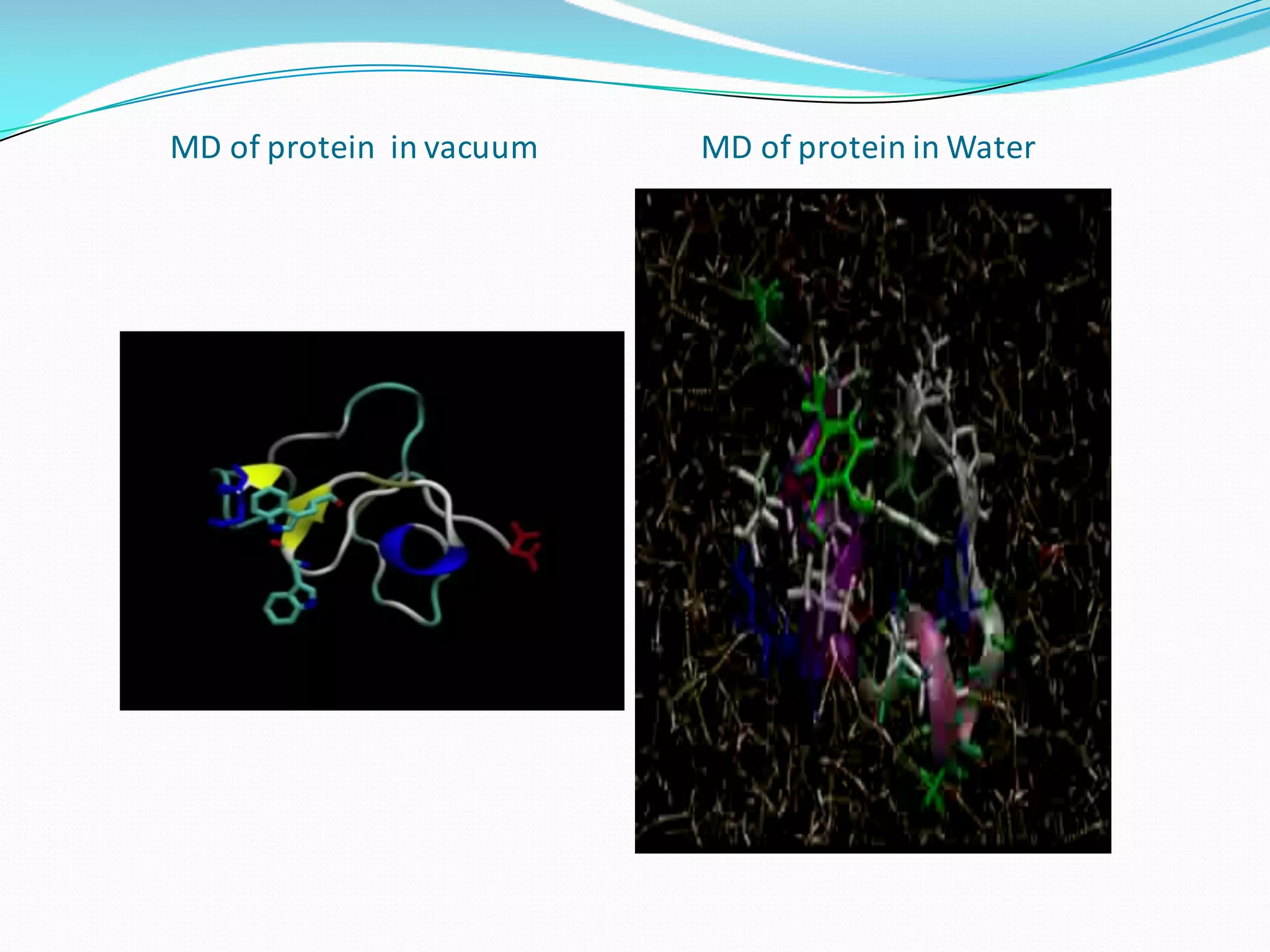


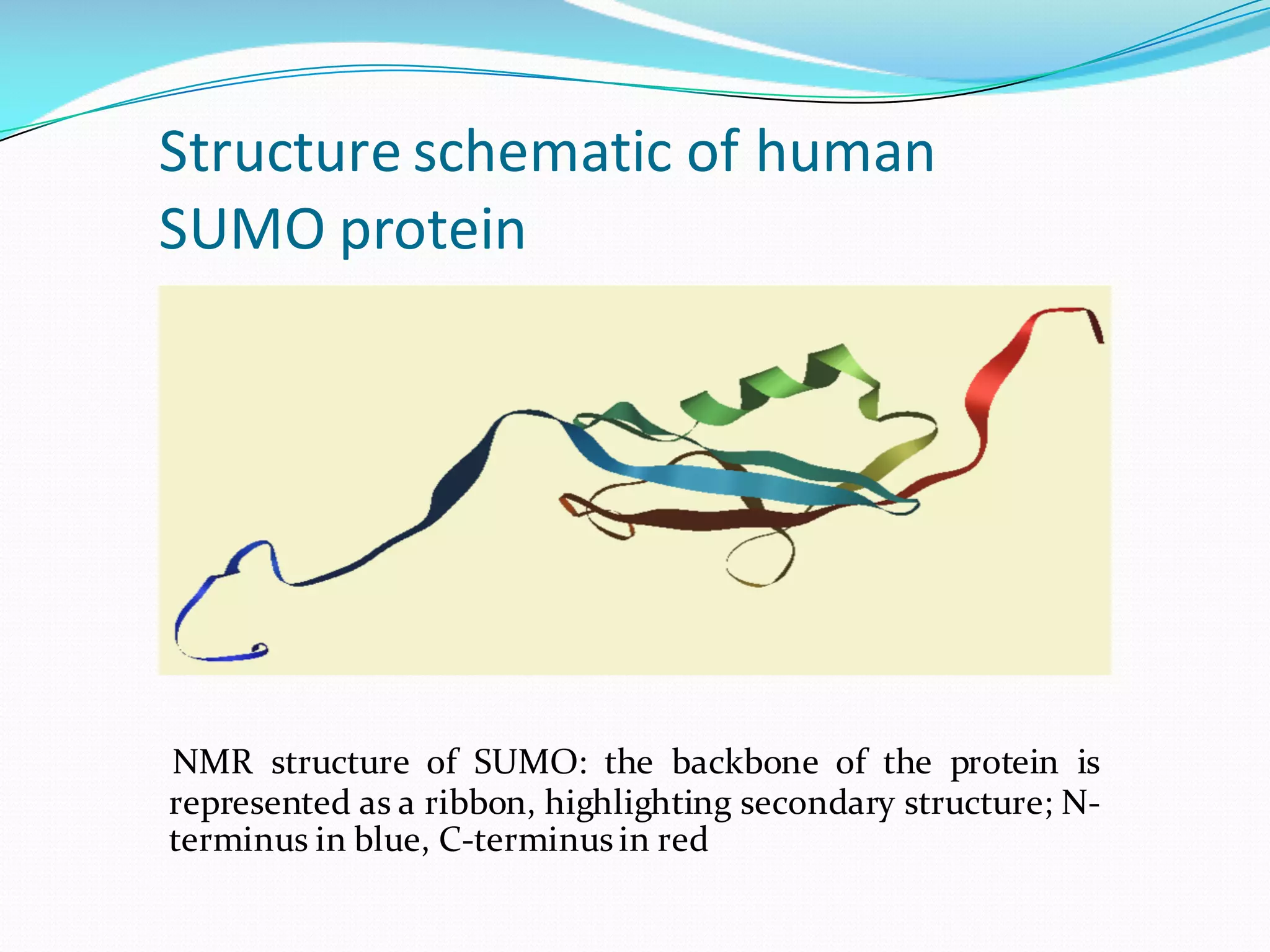












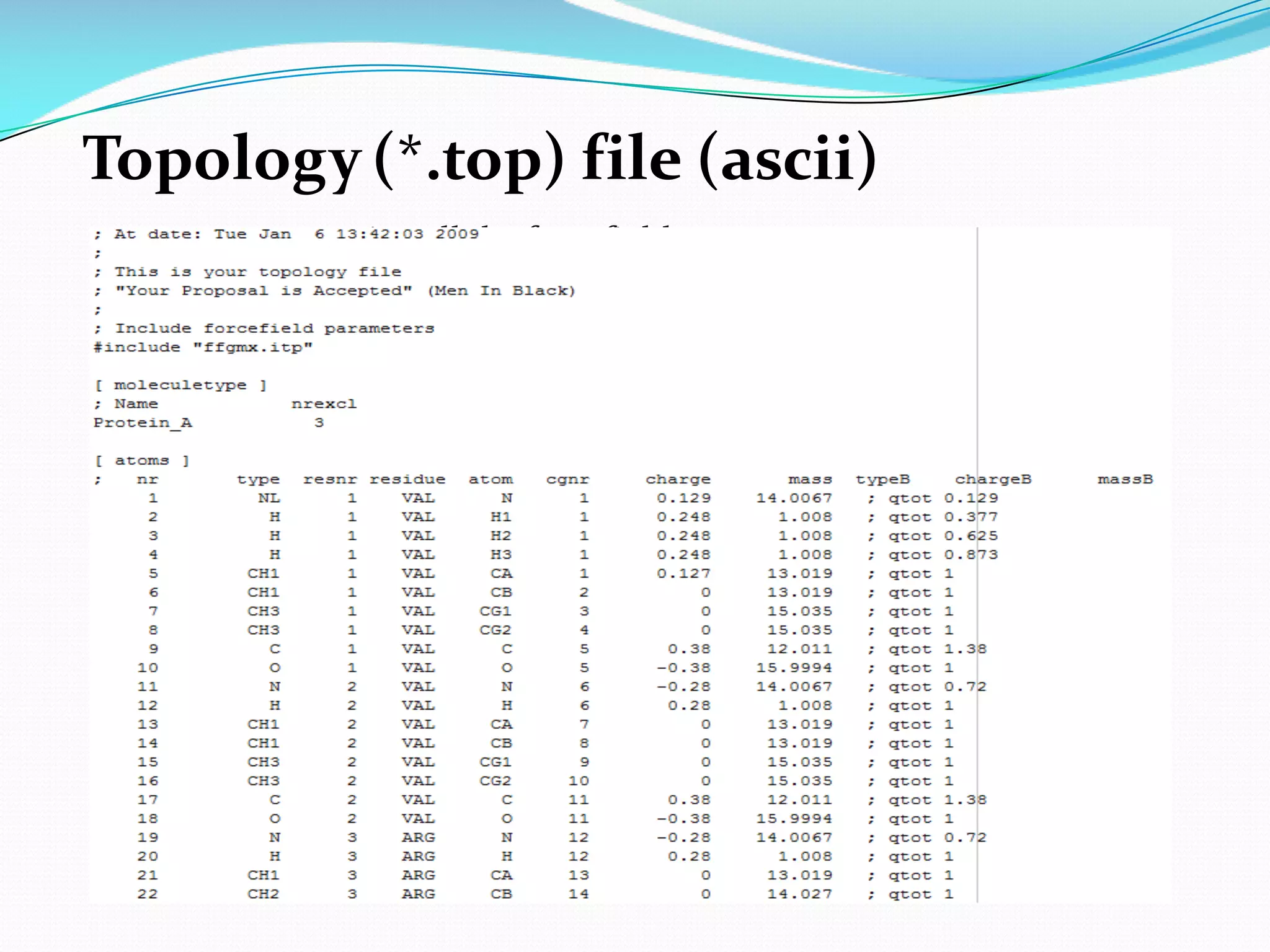


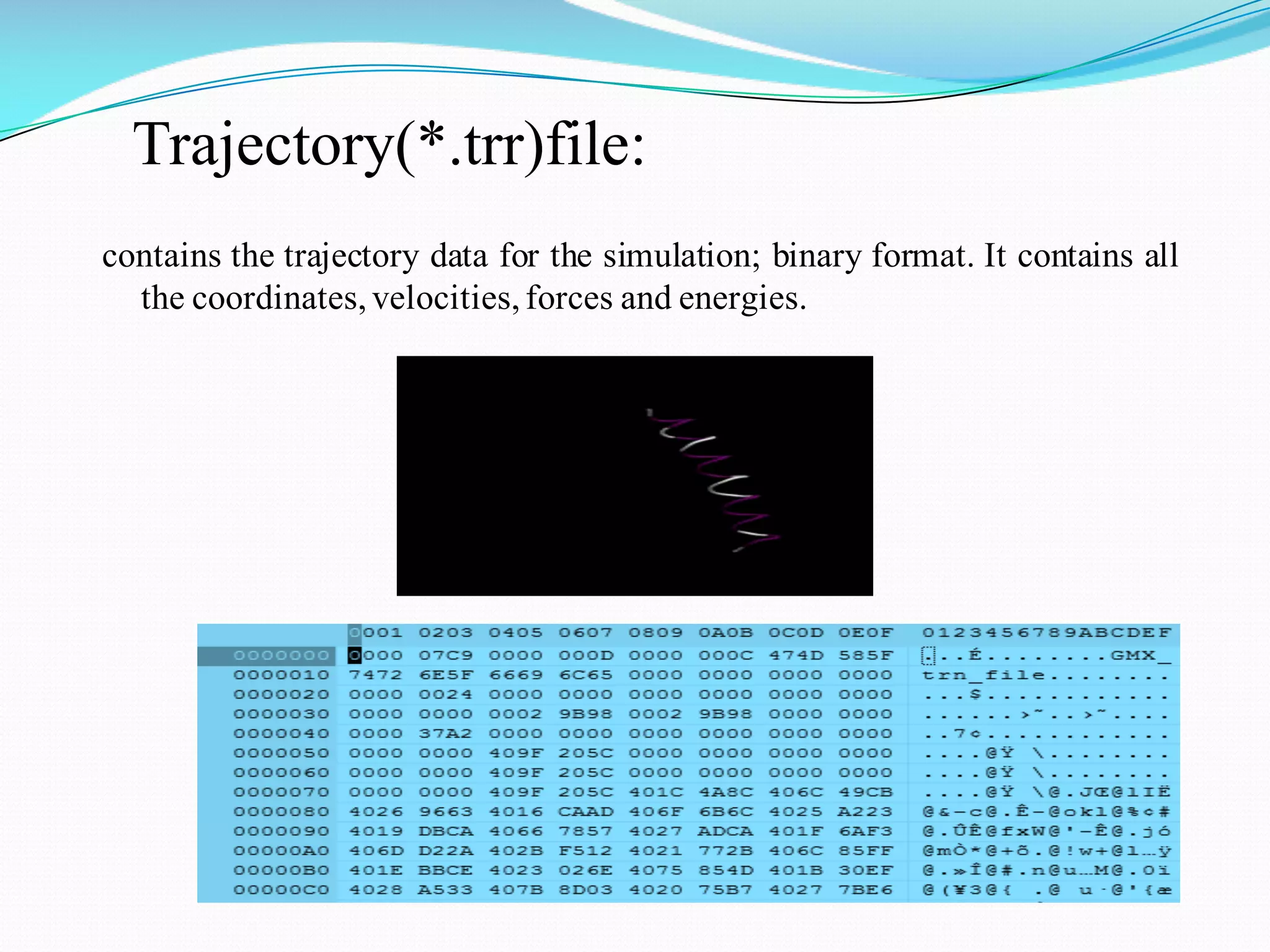


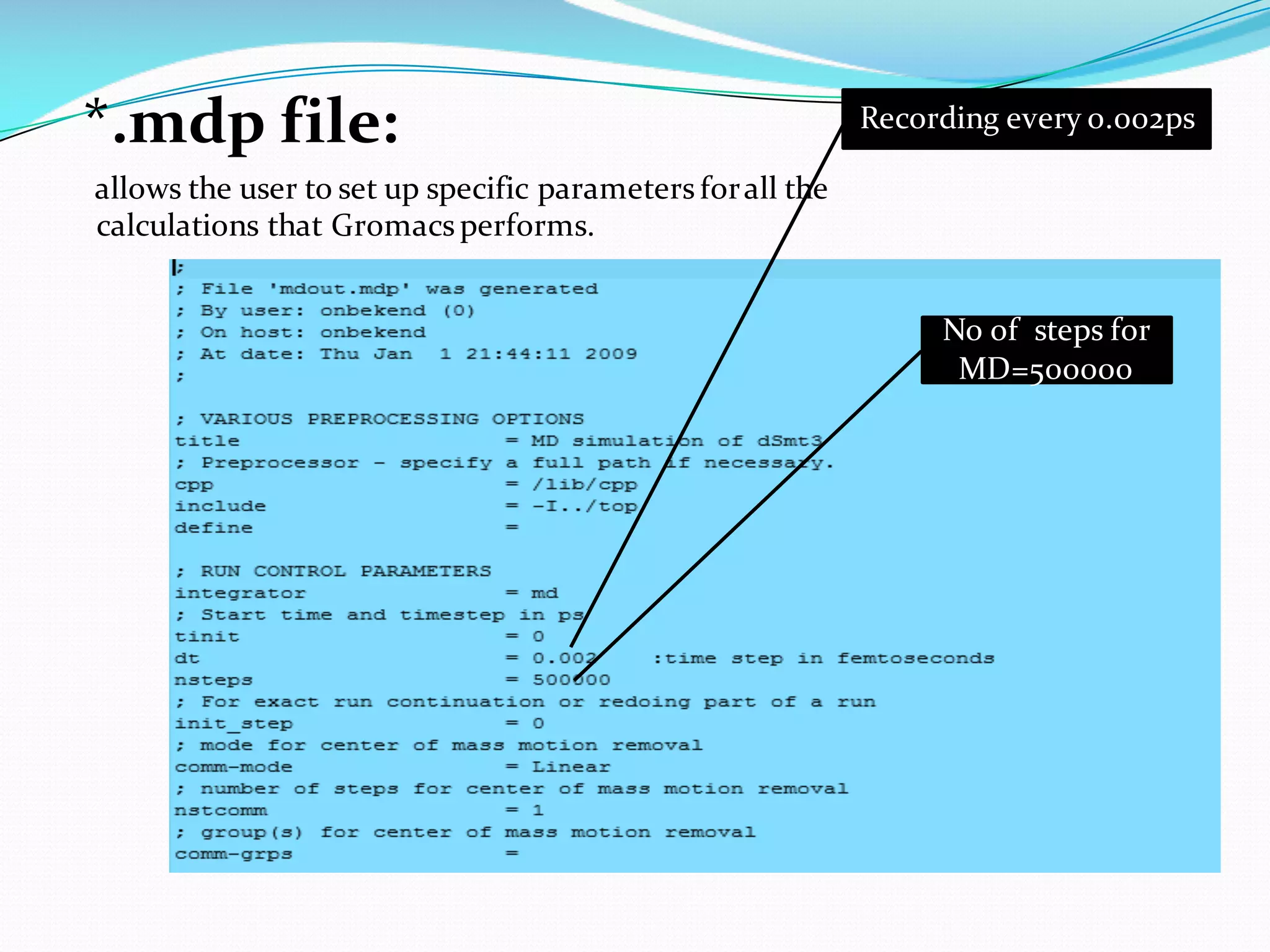
































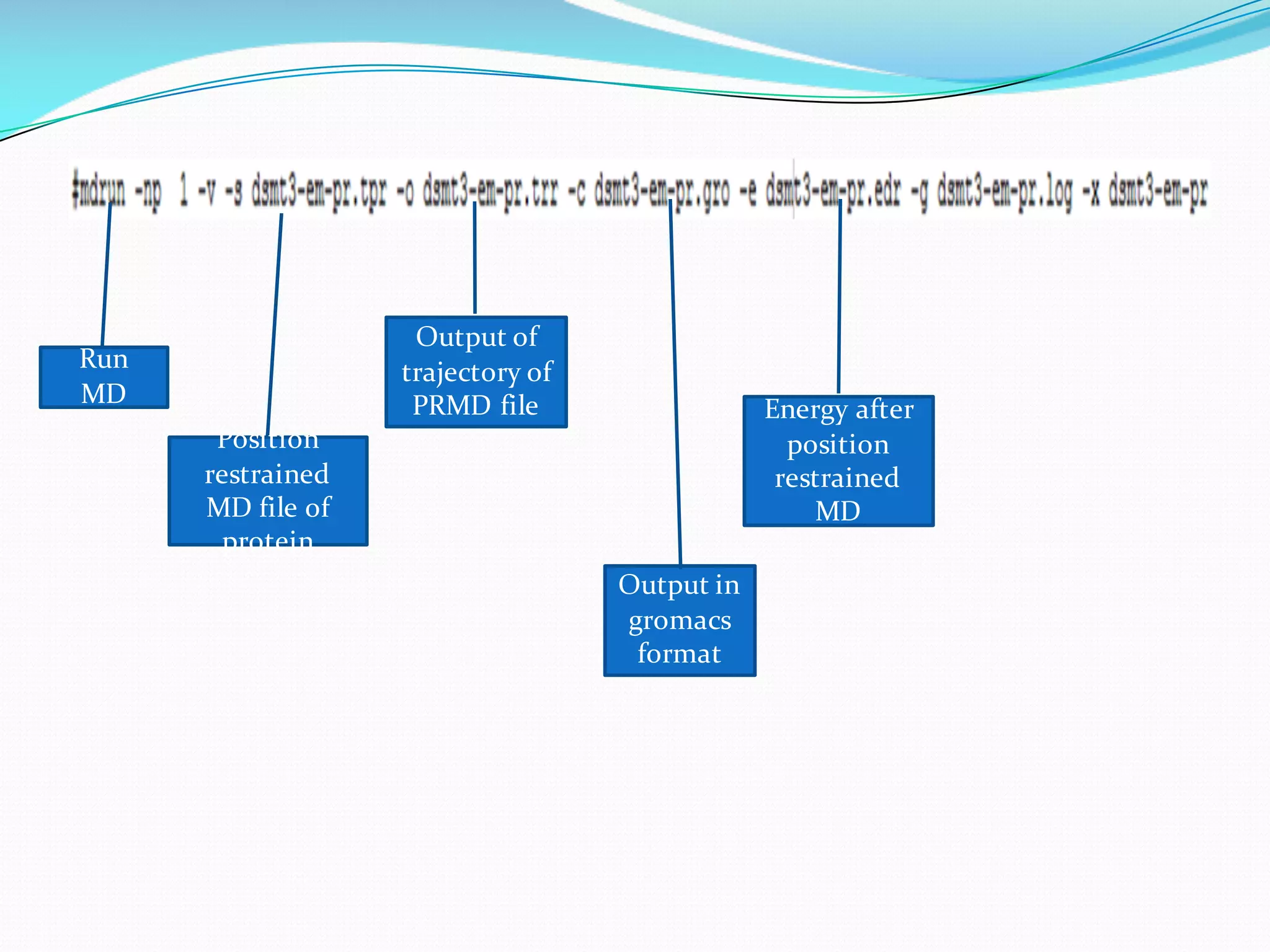








Molecular dynamics (MD) simulations allow atoms and molecules to interact over time, representing a virtual experiment. MD was used to give dynamics to SUMO proteins in solution. The SUMO protein was divided into fragments which were given random conformations using CYANA. These conformations were then converted to GROMACS format and molecular dynamics simulations were performed using GROMACS. The simulations involved energy minimization to relieve strain, followed by production runs. Various analysis tools were then used to analyze the results.















































