Downloaded 151 times






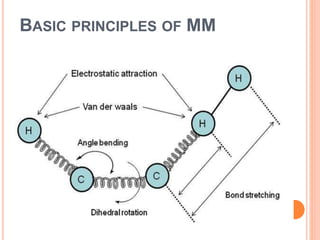



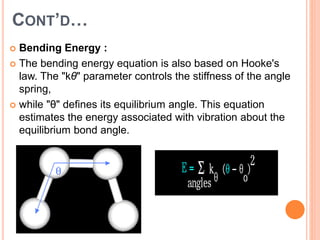



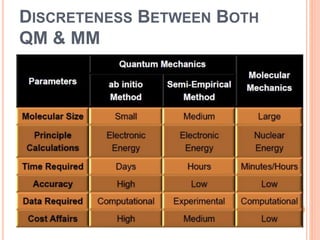
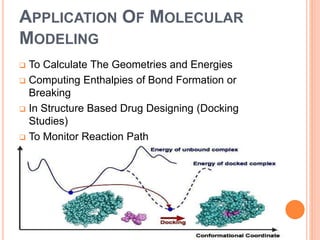



The document provides an overview of molecular mechanics as a computational method for studying molecular systems, focusing on potential energy surfaces and the calculations of various energy components including bonded and non-bonded interactions. It outlines fundamental principles of molecular mechanics, the role of force fields, and various applications in drug design and simulation. Additionally, it mentions novel techniques that combine quantum mechanics with molecular mechanics for enhanced accuracy in modeling complex systems.



























































![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)







