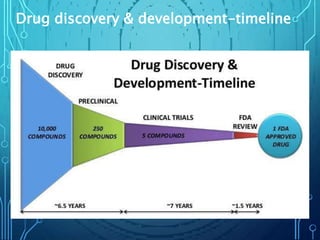
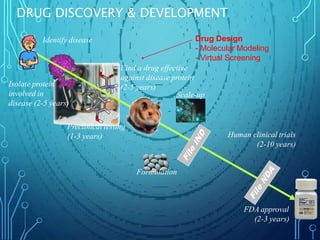
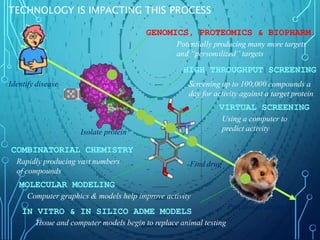
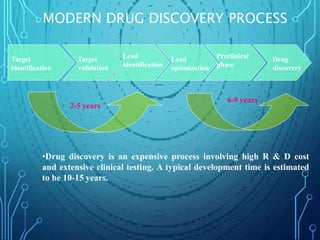
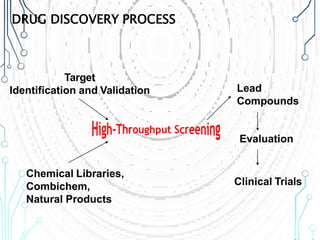
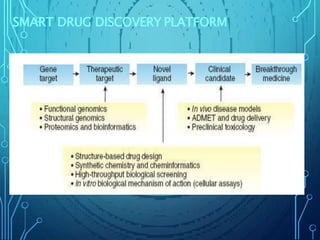
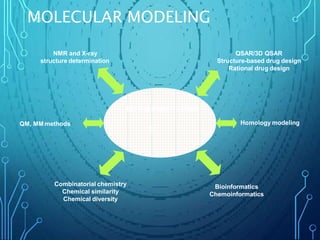
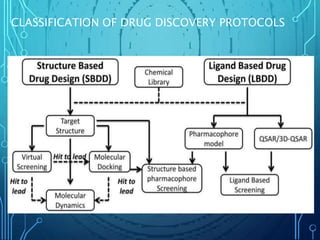
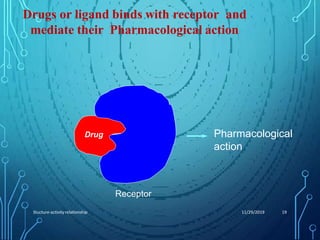
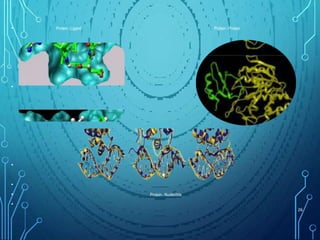
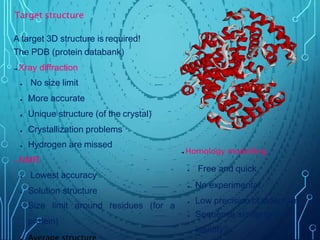
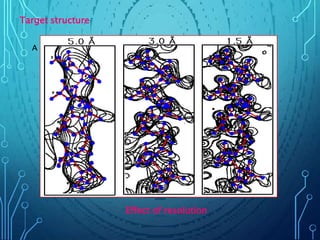
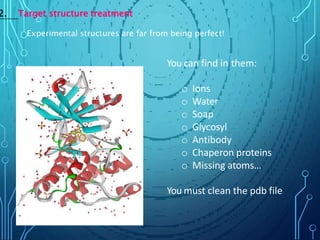
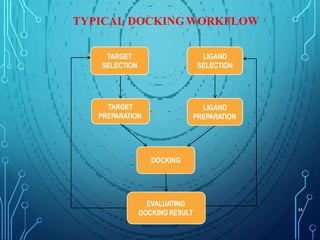
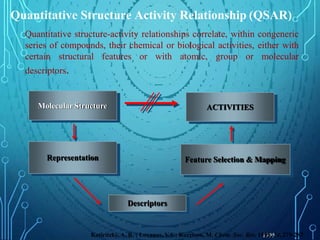
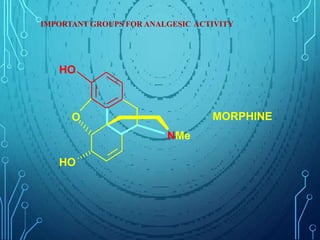
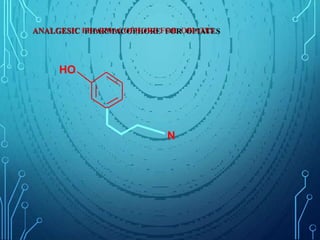
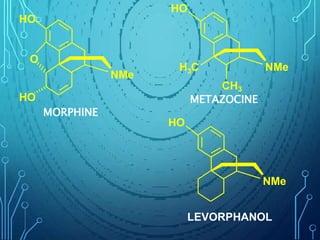
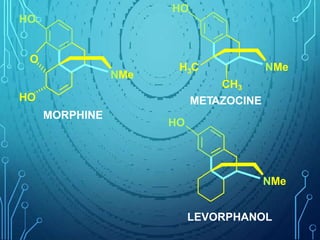
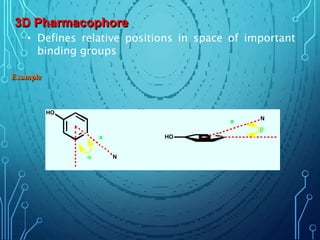
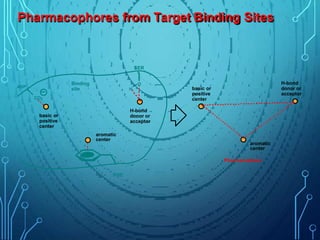
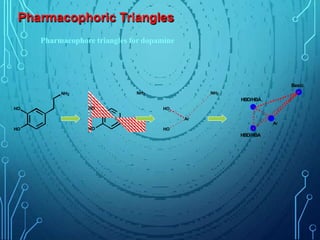
Drug discovery is an expensive and lengthy process involving high costs and extensive testing over 10-15 years. Computer-aided drug design techniques like molecular modeling, virtual screening, and quantitative structure-activity relationships (QSAR) are helping to improve the drug discovery process. Molecular docking uses computer models to predict how drug molecules bind to their protein targets. Key steps in docking include target and ligand preparation, docking simulations, and analysis of results. Factors like intermolecular forces, flexibility, and binding site selection influence docking accuracy. QSAR analyses seek mathematical correlations between compound structures and their biological activities to enable prediction of new candidates.














































![DRUG DESIGN SUCCESSES
While we are still waiting for a drug totally designed from
scratch, many drugs have been developed with major
contributions from computational methods
norfloxacin (1983)
antibiotic
first of the 6-fluoroquinolones
QSAR studies
dorzolamide [Trusopt] (1994)
glaucoma treatment
carbonic anhydrase inhibitor
SBLD and ab initio calcs
donepezil (1996)
Alzheimer's treatment
acetylcholinesterase inhibitor
shape analysis and docking studies
losartan [Cozaar] (1995)
angiotensin II antagonist
anti-hypertensive
Modeling Angiotensin II octapeptide
zolmatriptan [Zomig] 1995
5-HT1D agonist
migraine treatment
Molecular modeling](https://image.slidesharecdn.com/cadd-1911291340501-221116114705-8a399c1b/85/cadd-191129134050-1-pptx-79-320.jpg)

























































































