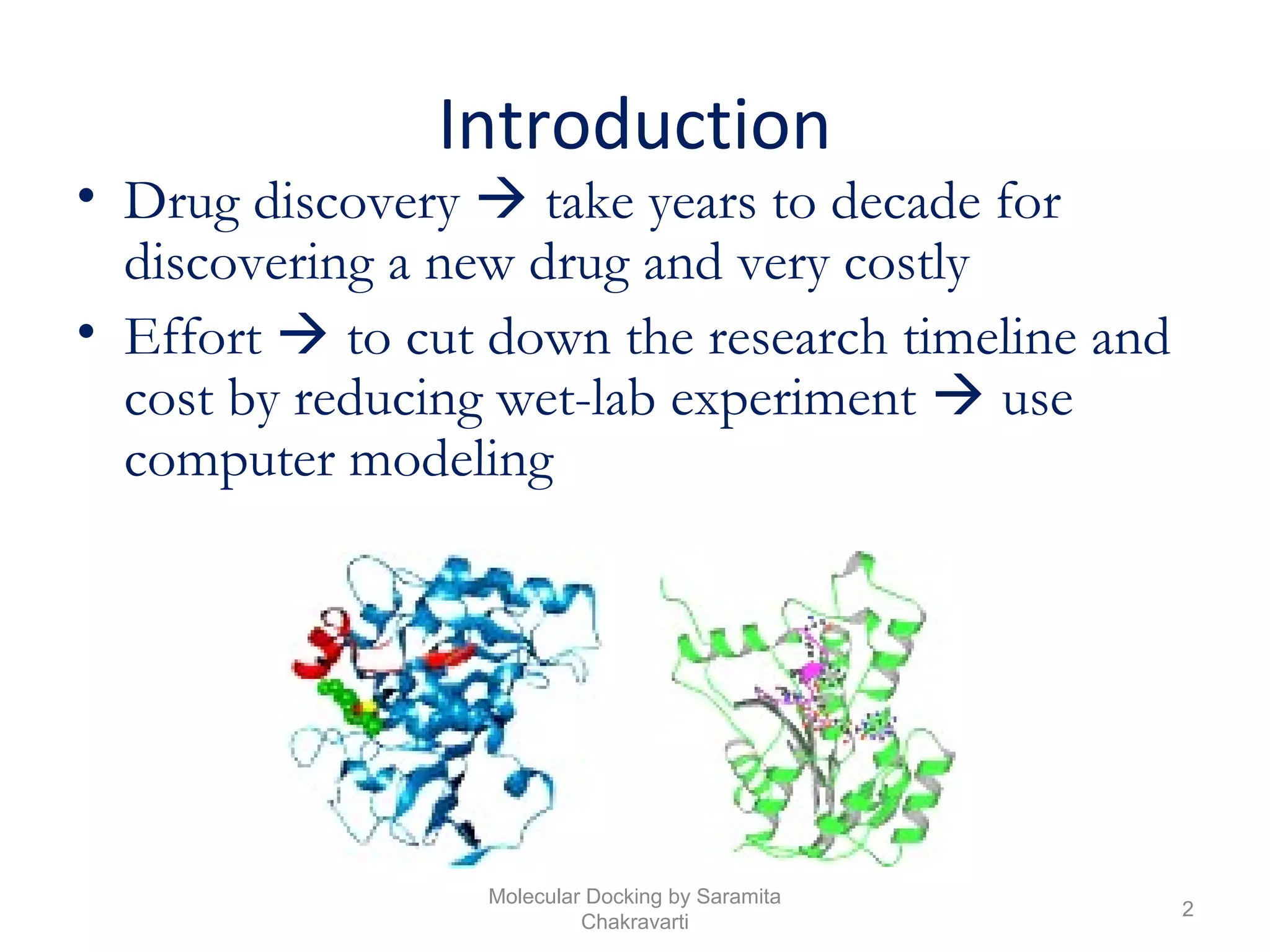
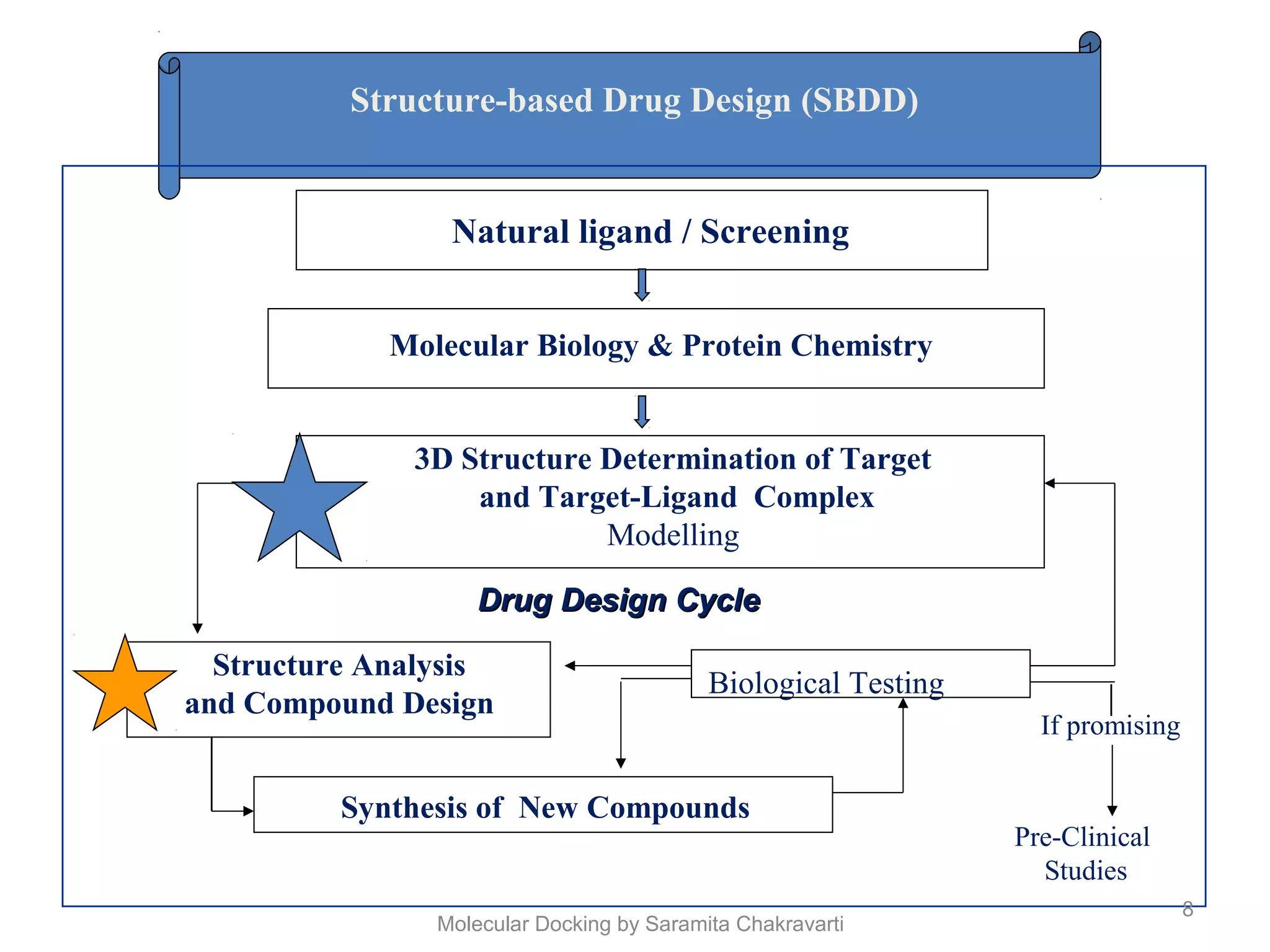
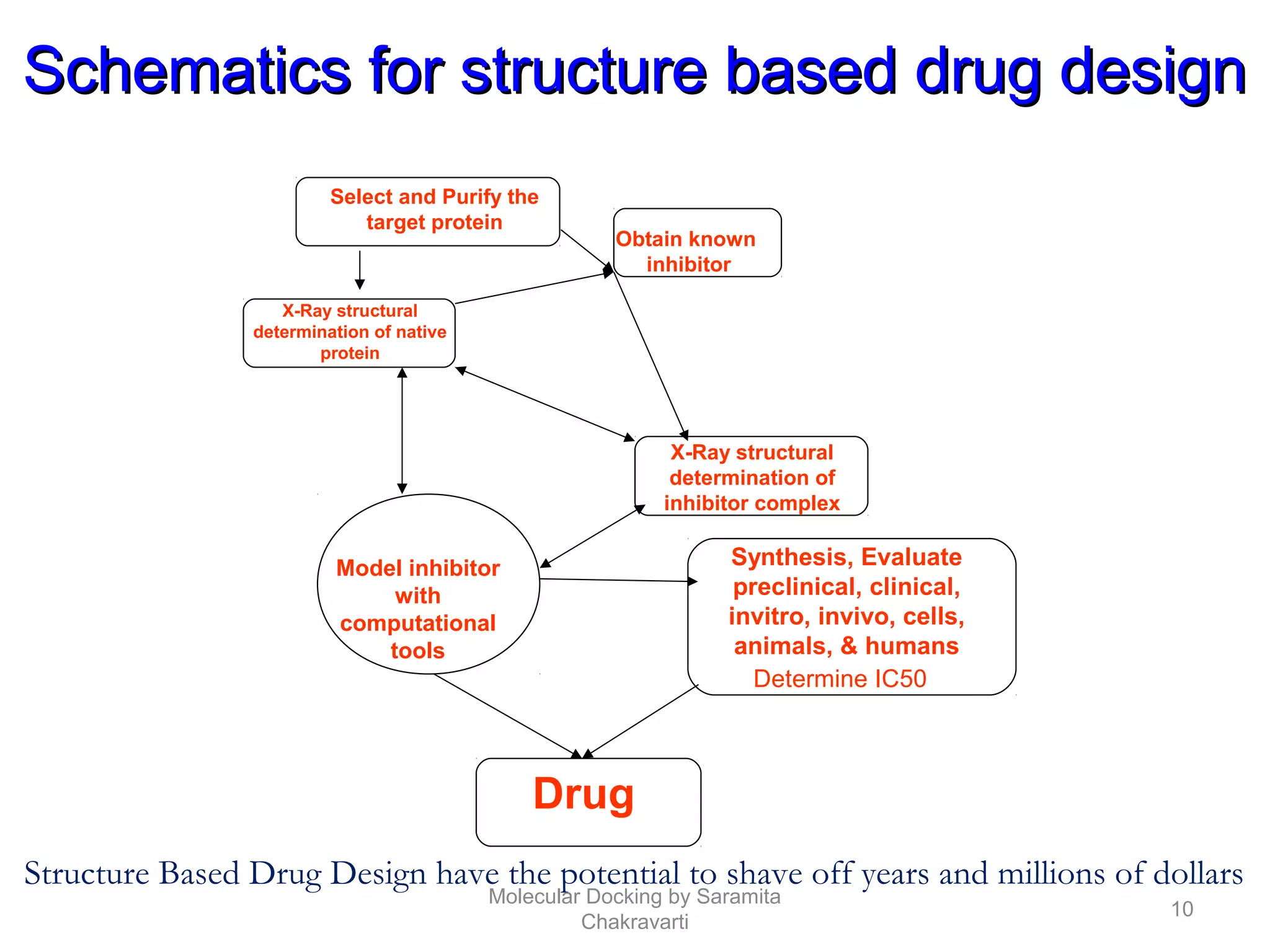
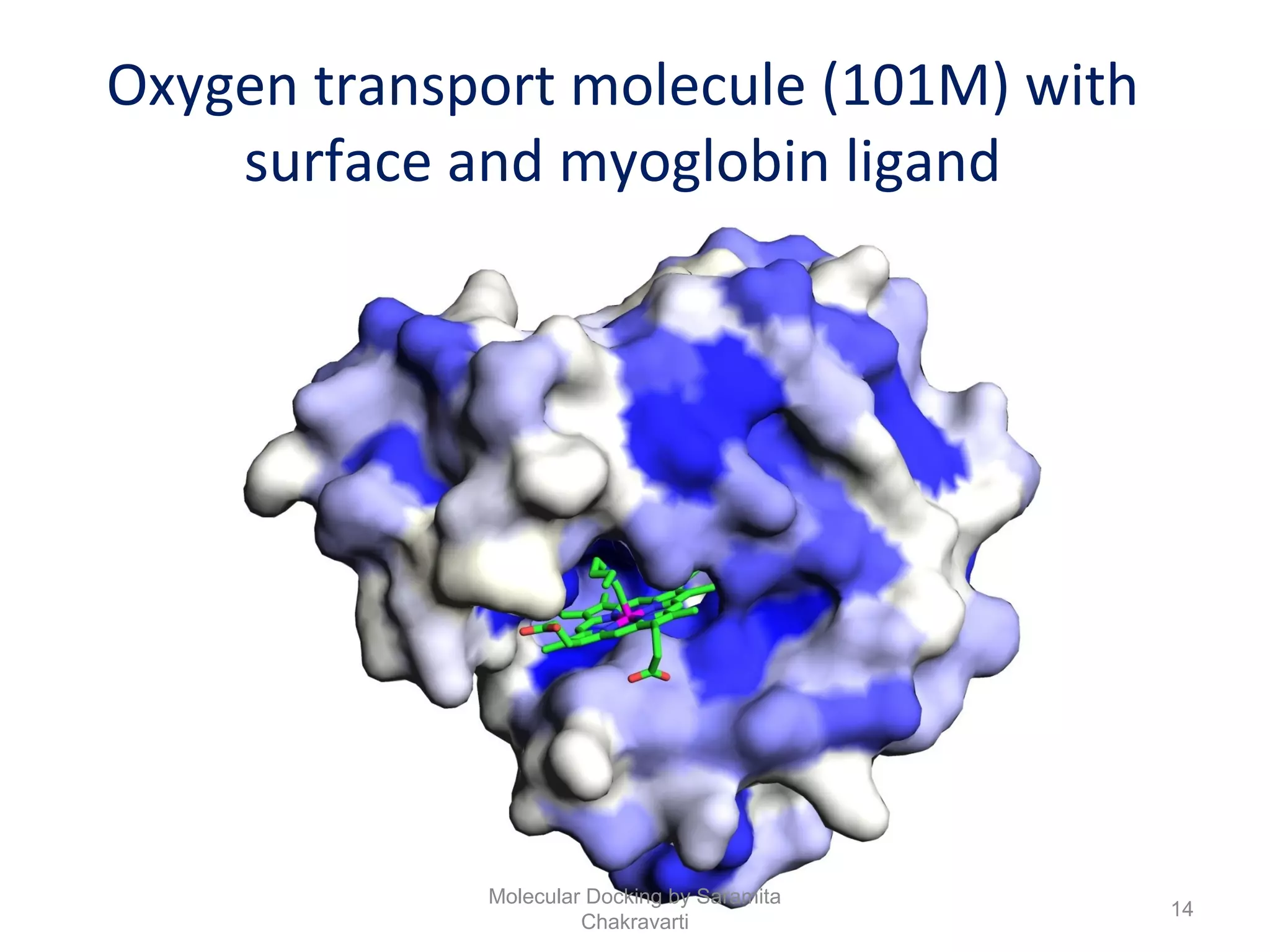
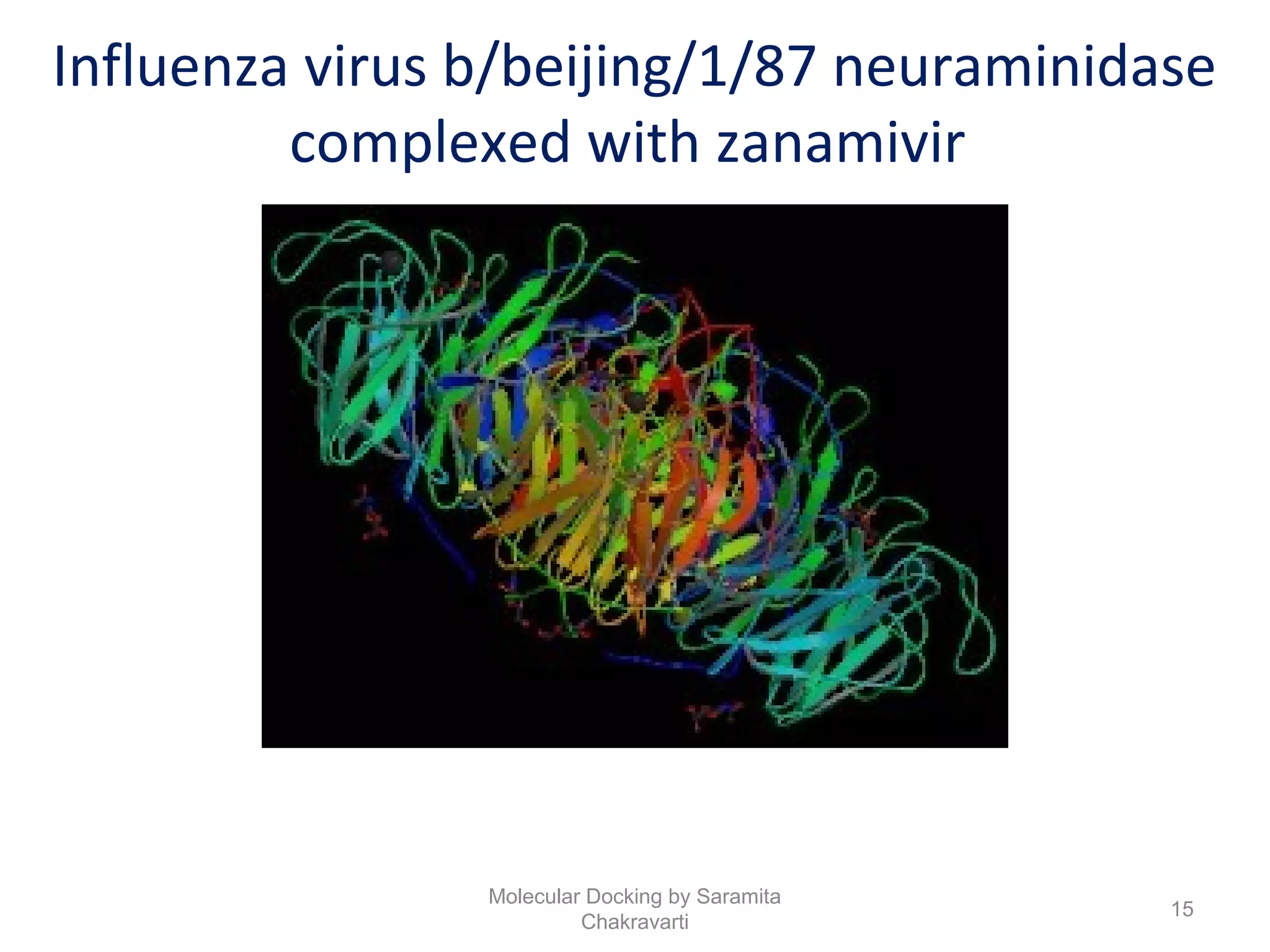
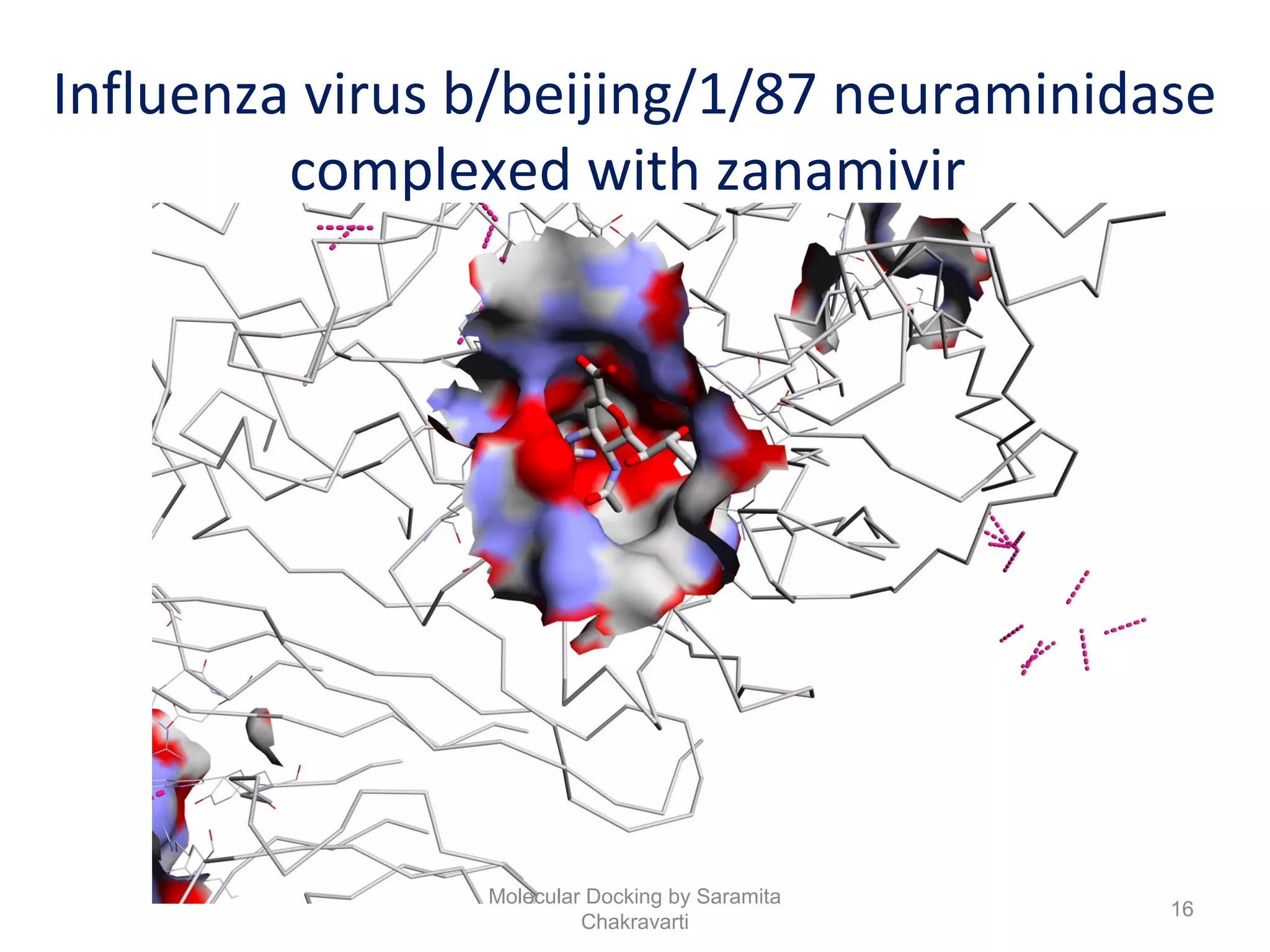
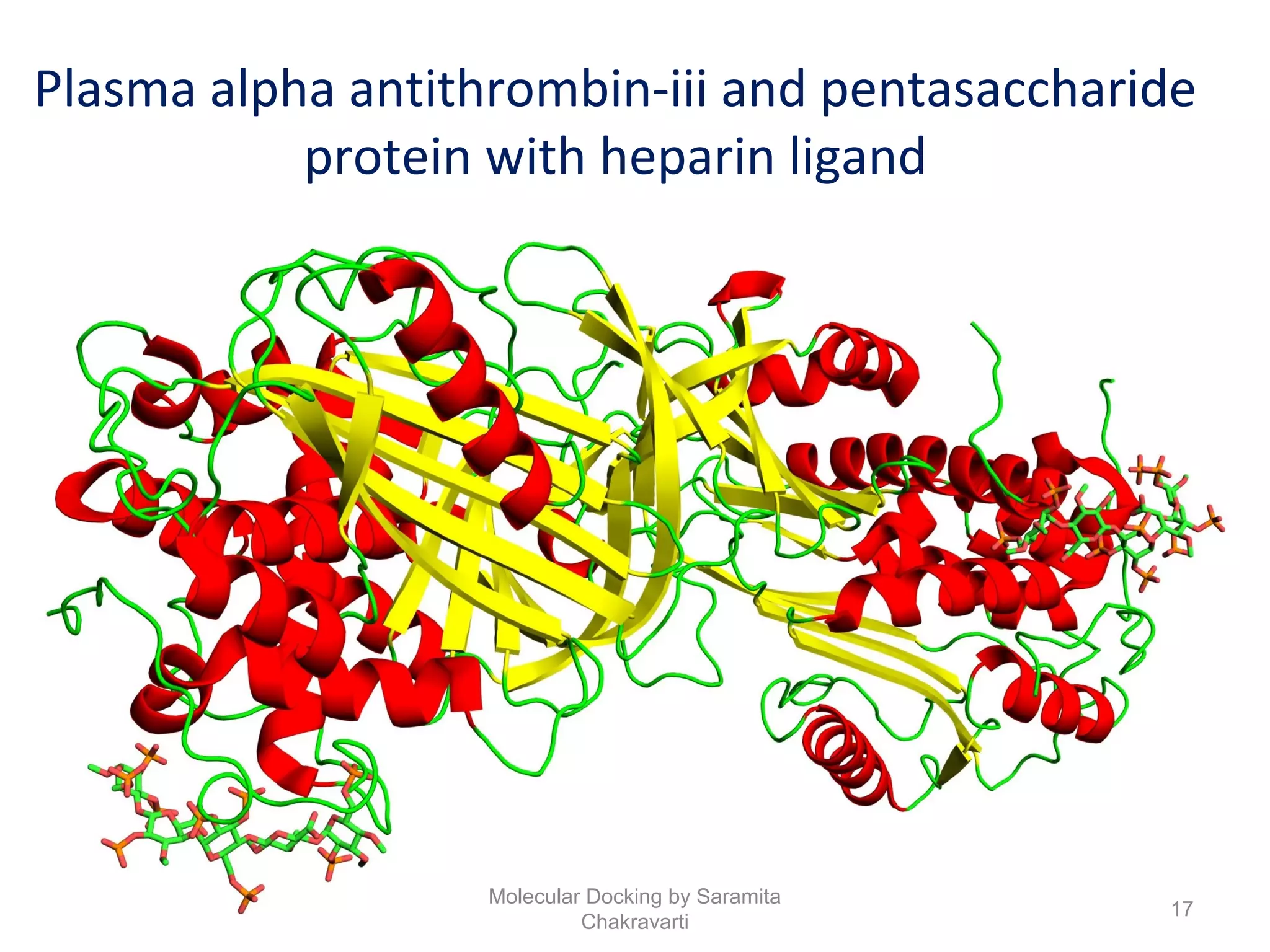
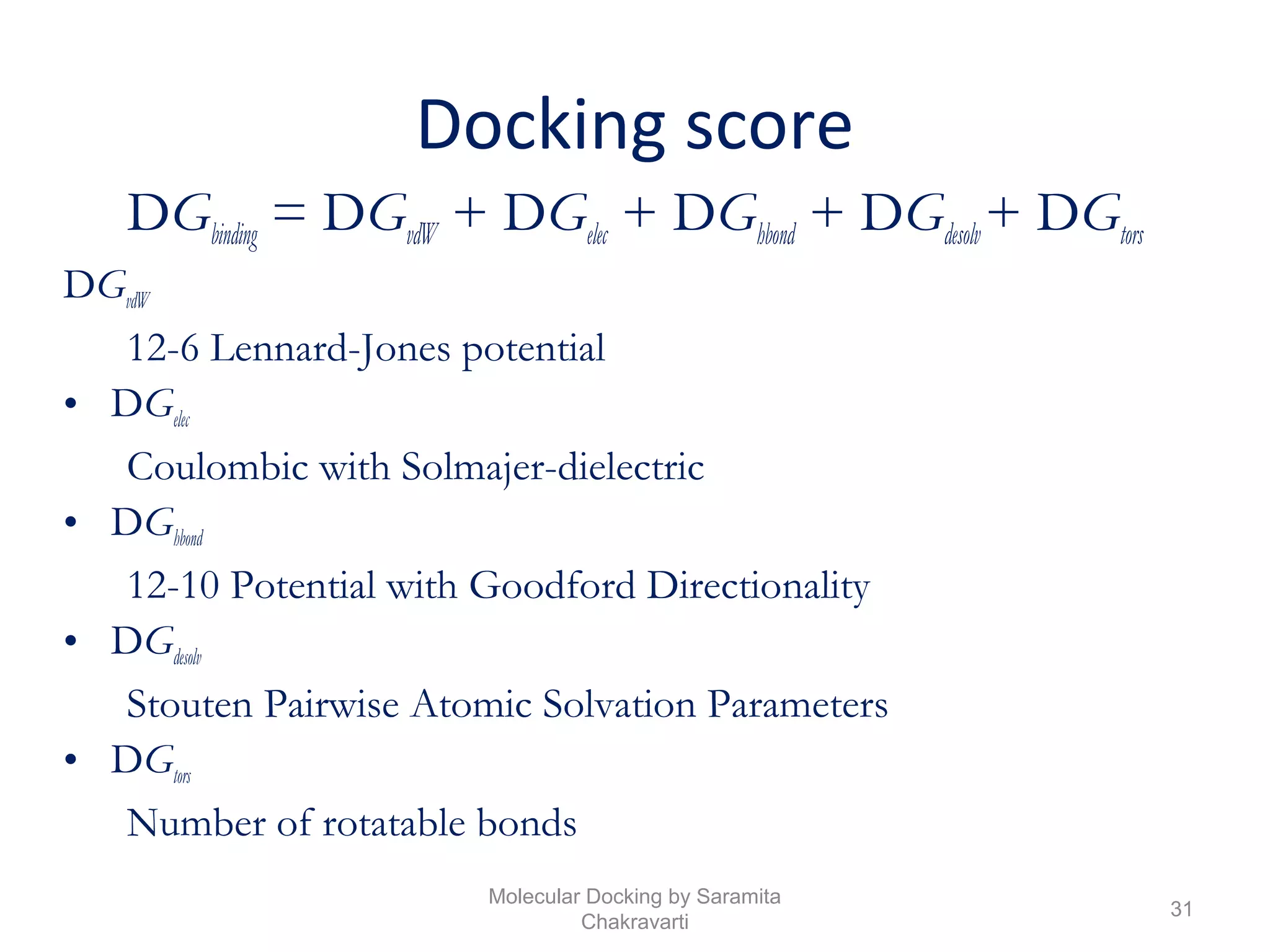
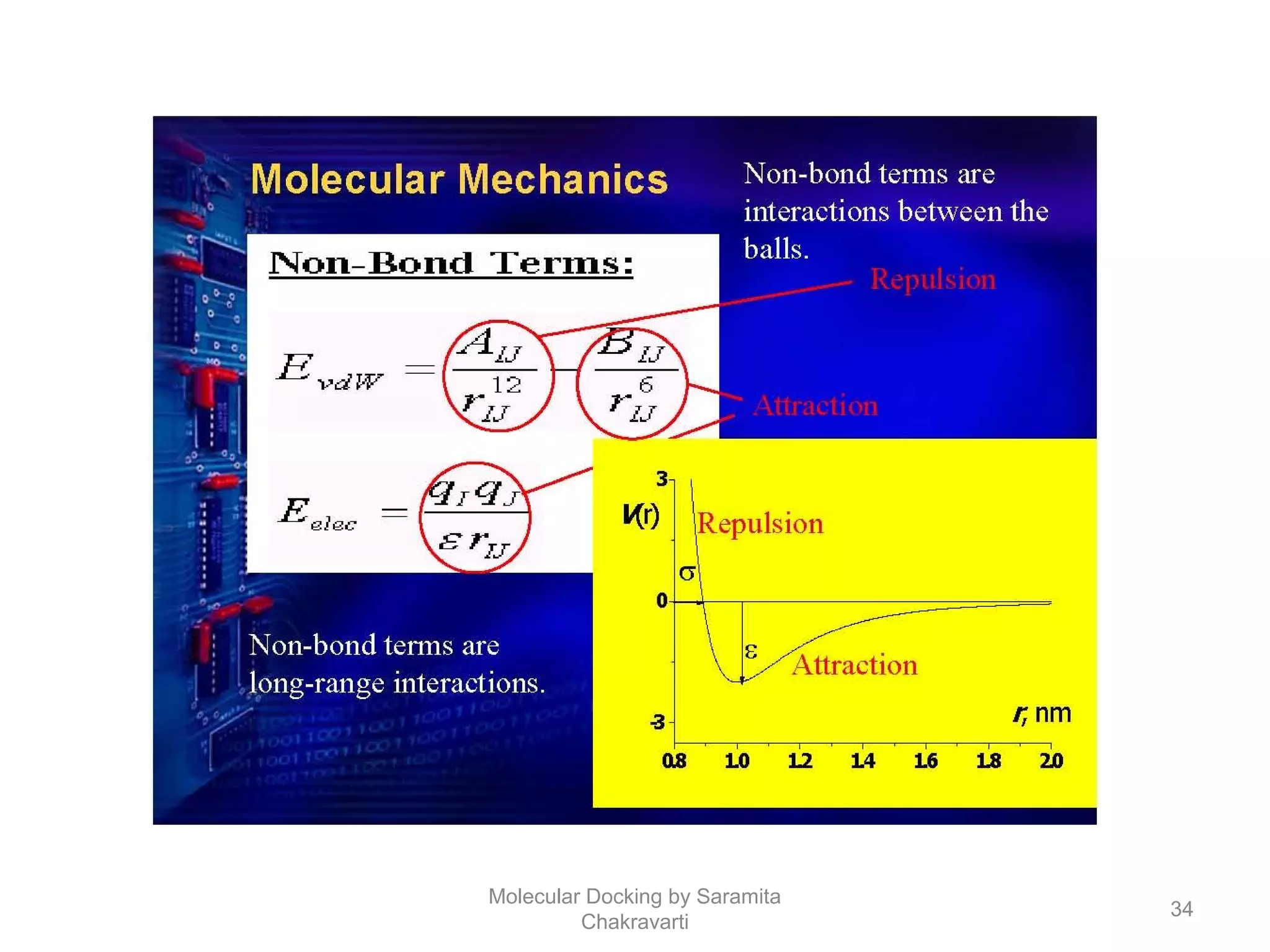
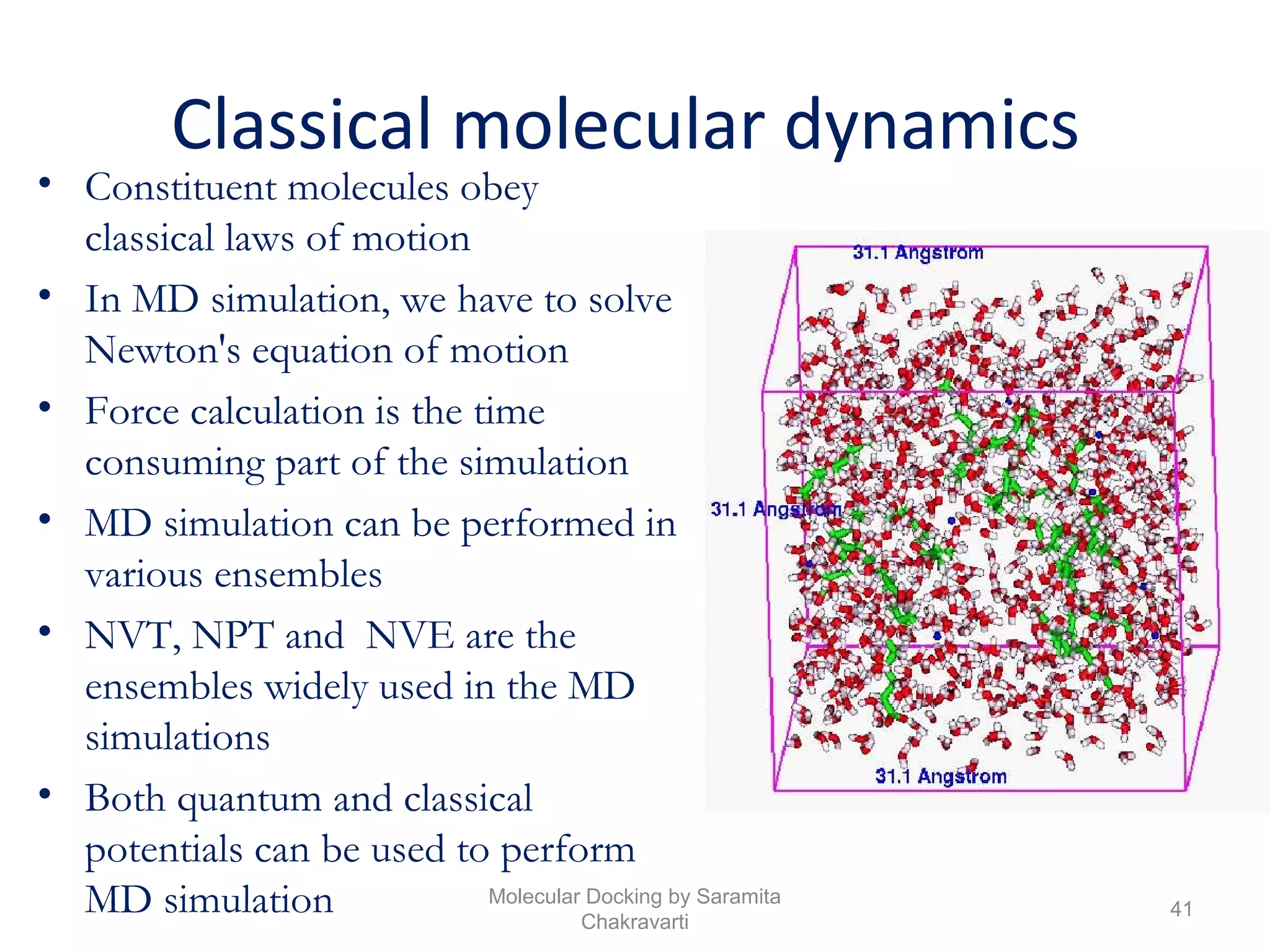
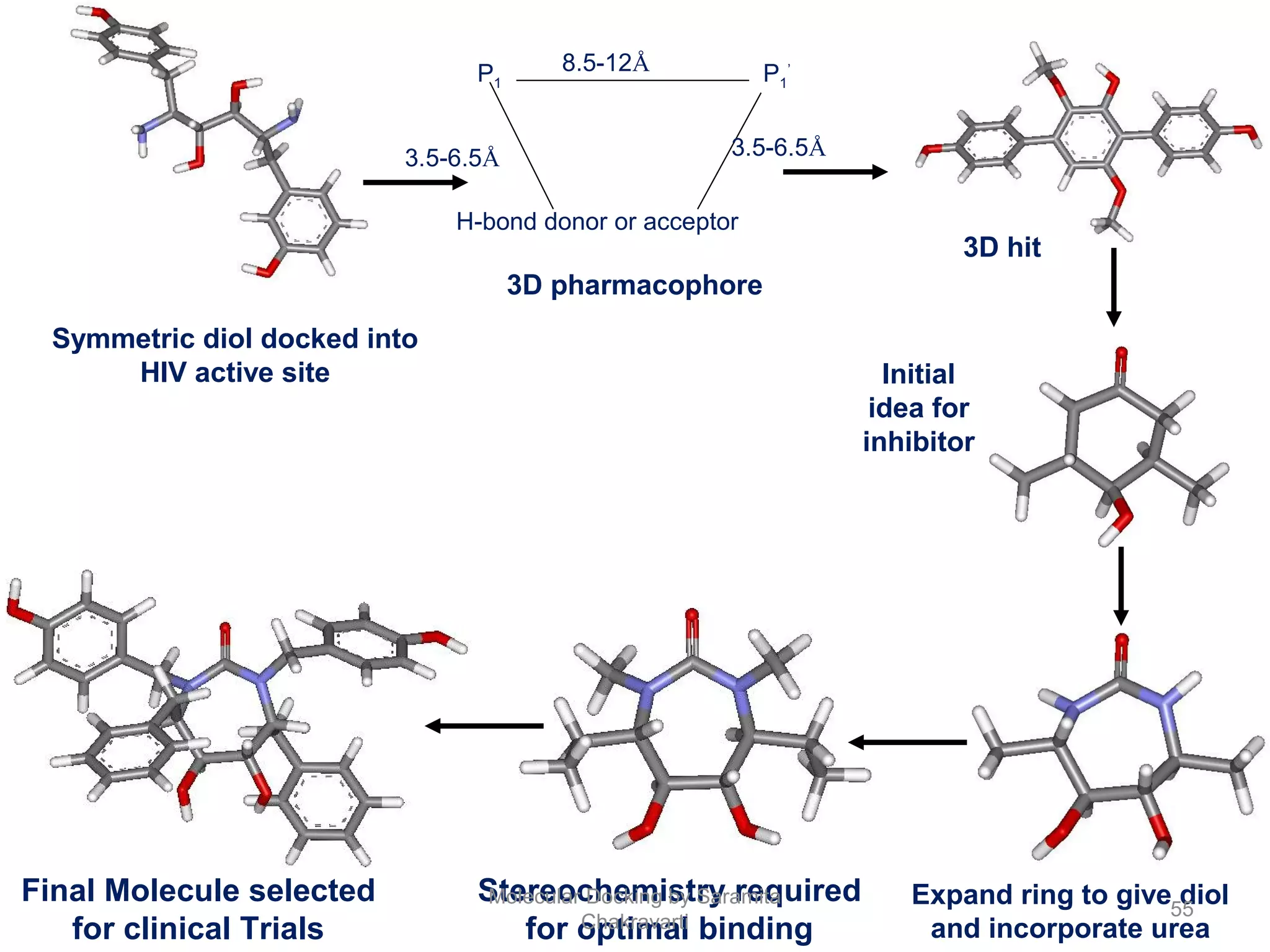
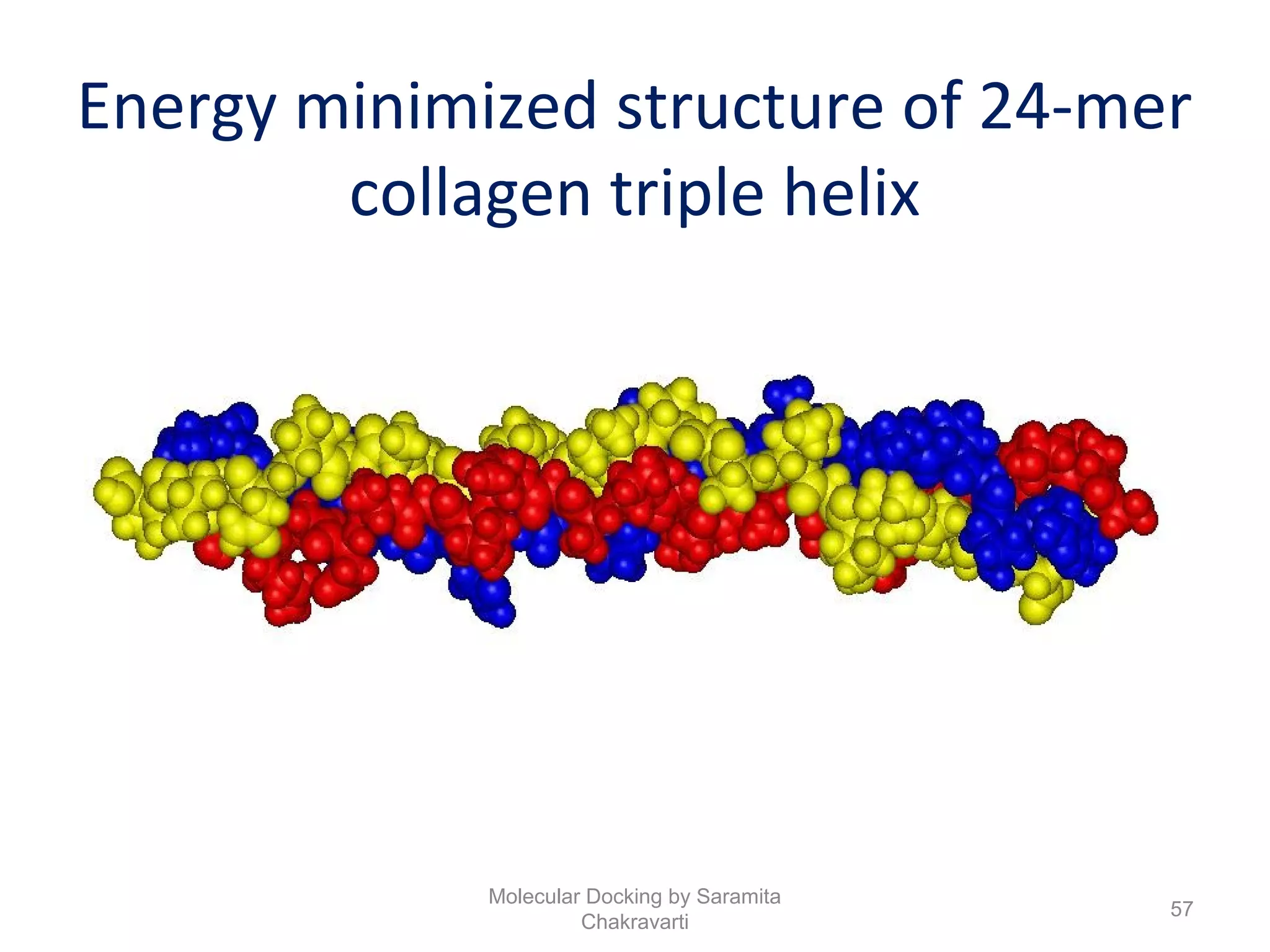
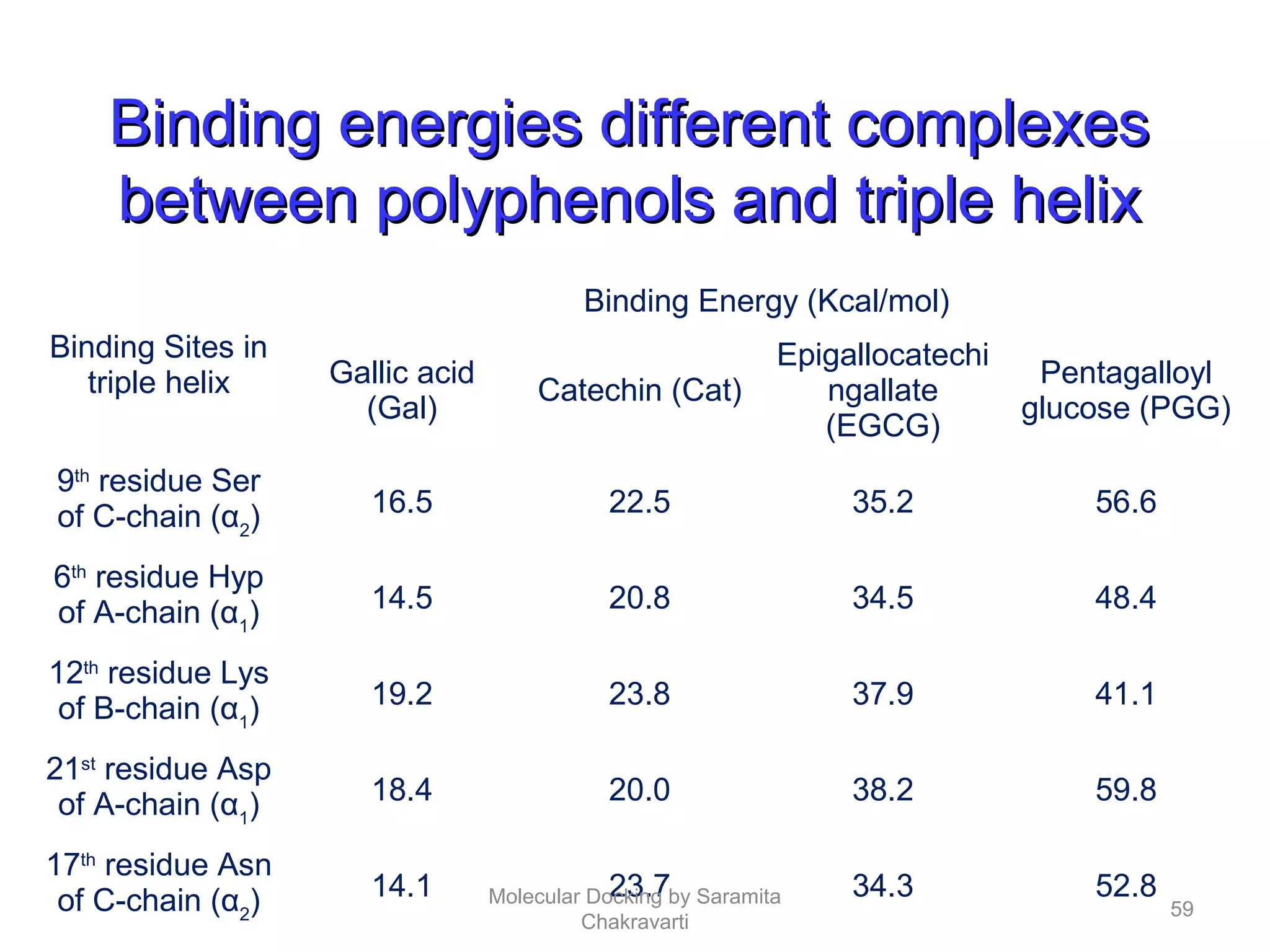
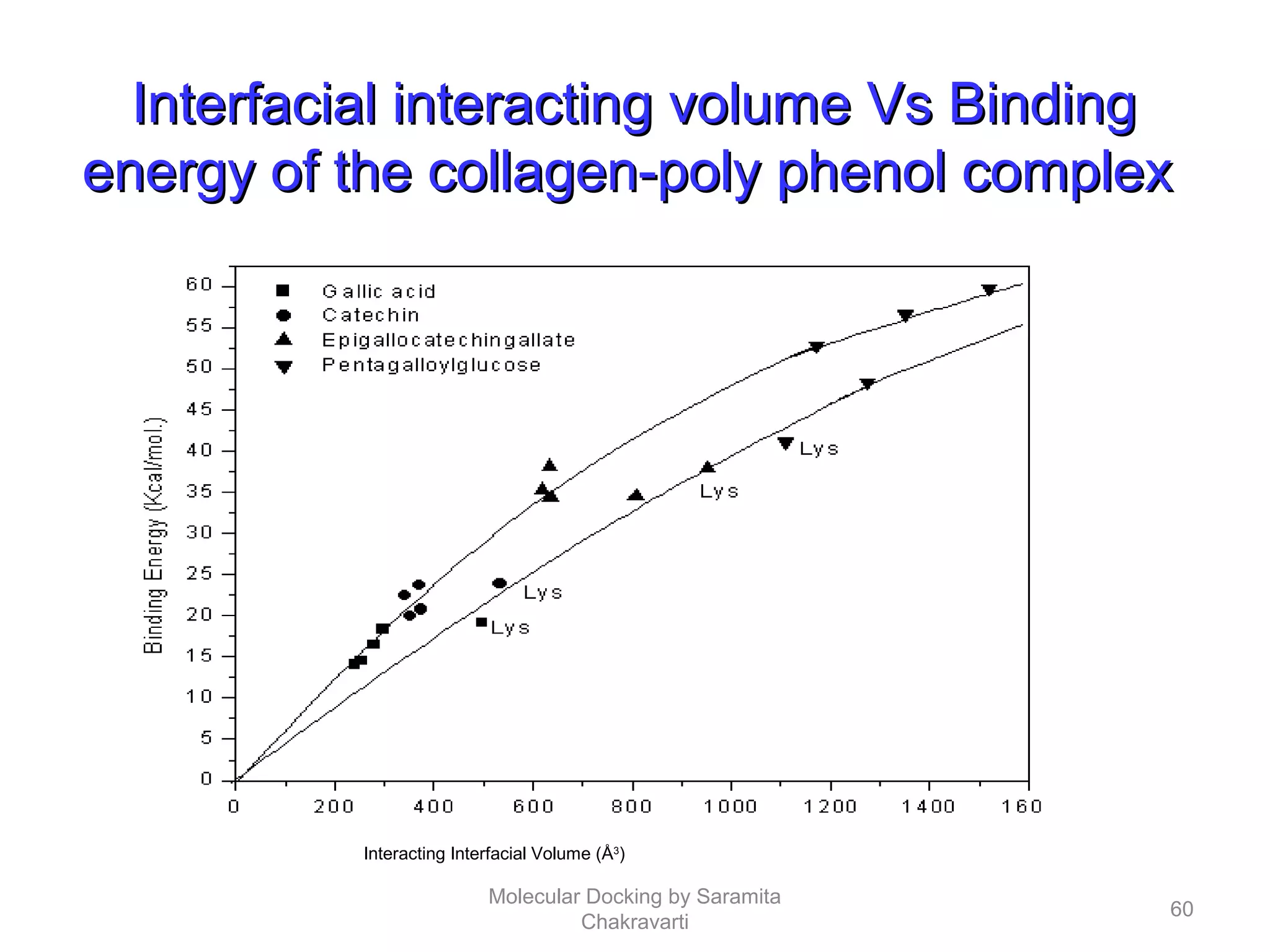
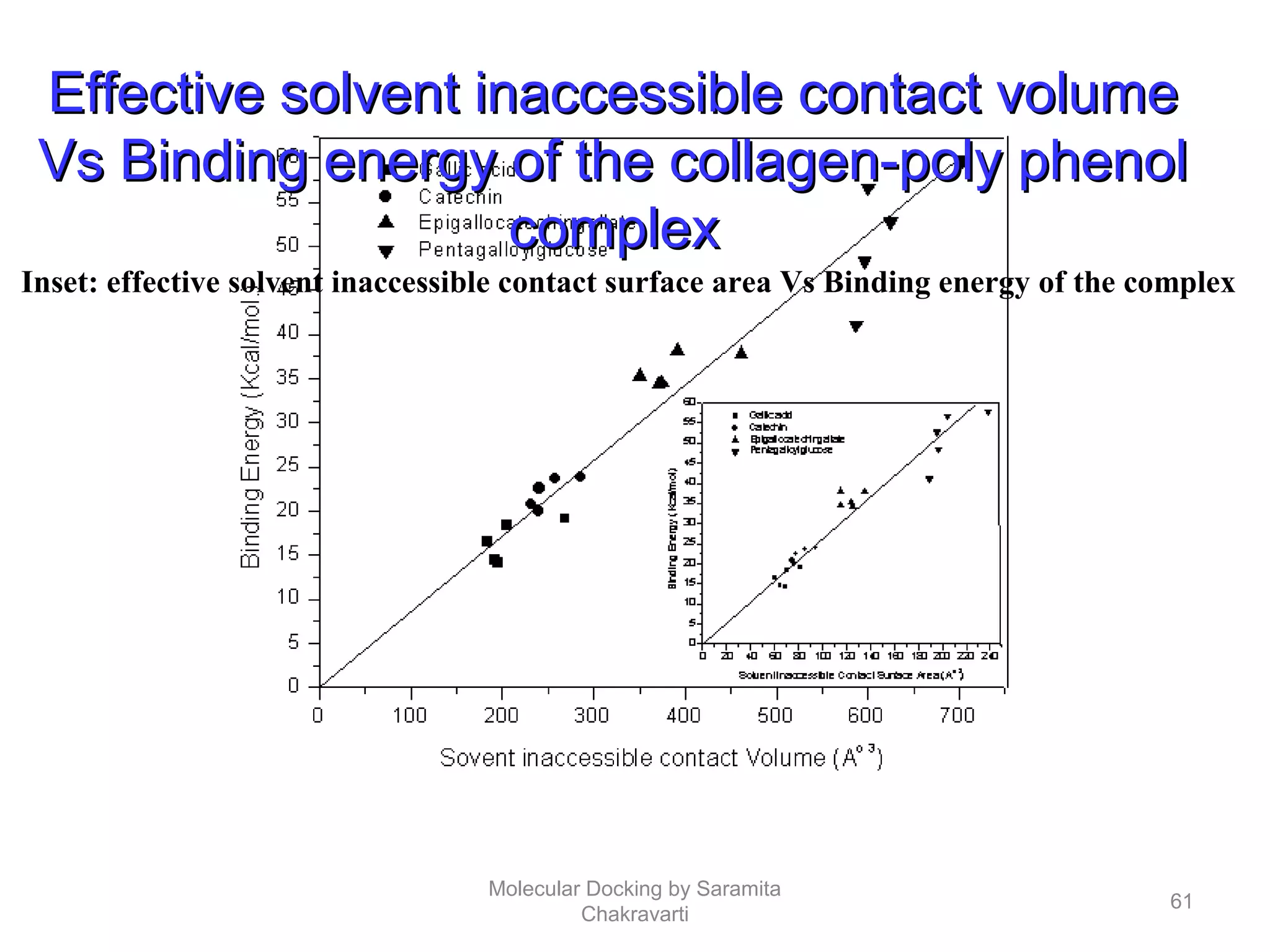
The document discusses molecular docking as a computational approach in drug discovery, aiming to reduce time and costs associated with wet-lab experiments. It covers methods for identifying lead compounds, structure-based drug design, and the use of molecular modeling techniques in predicting receptor-ligand interactions. Additionally, it highlights the importance of search methods and scoring functions in molecular docking processes.