An overview ofthe drug discovery process
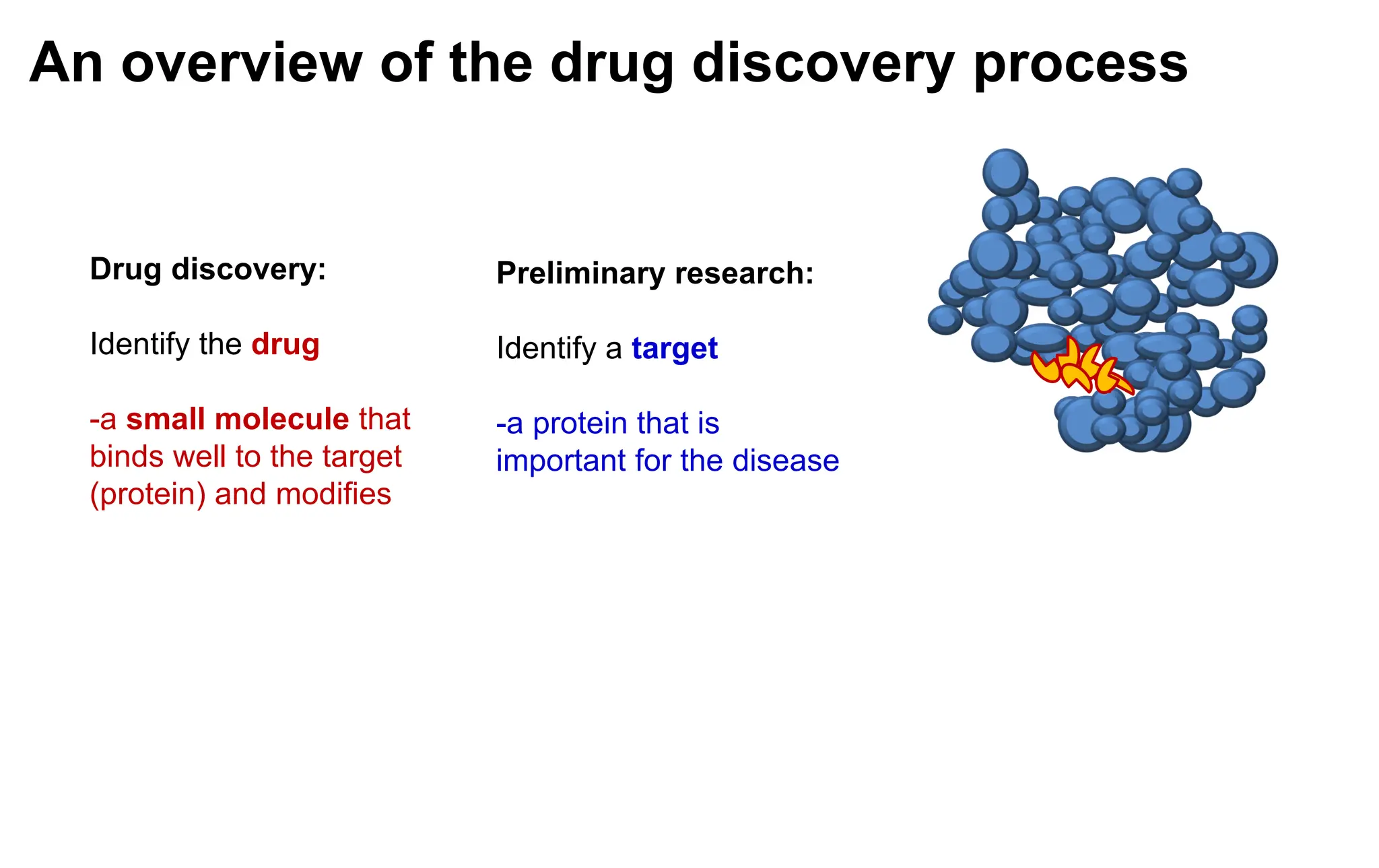
Preliminary research:
Identify a target
-a protein that is
important for the disease
Drug discovery:
Identify the drug
-a small molecule that
binds well to the target
(protein) and modifies
3.
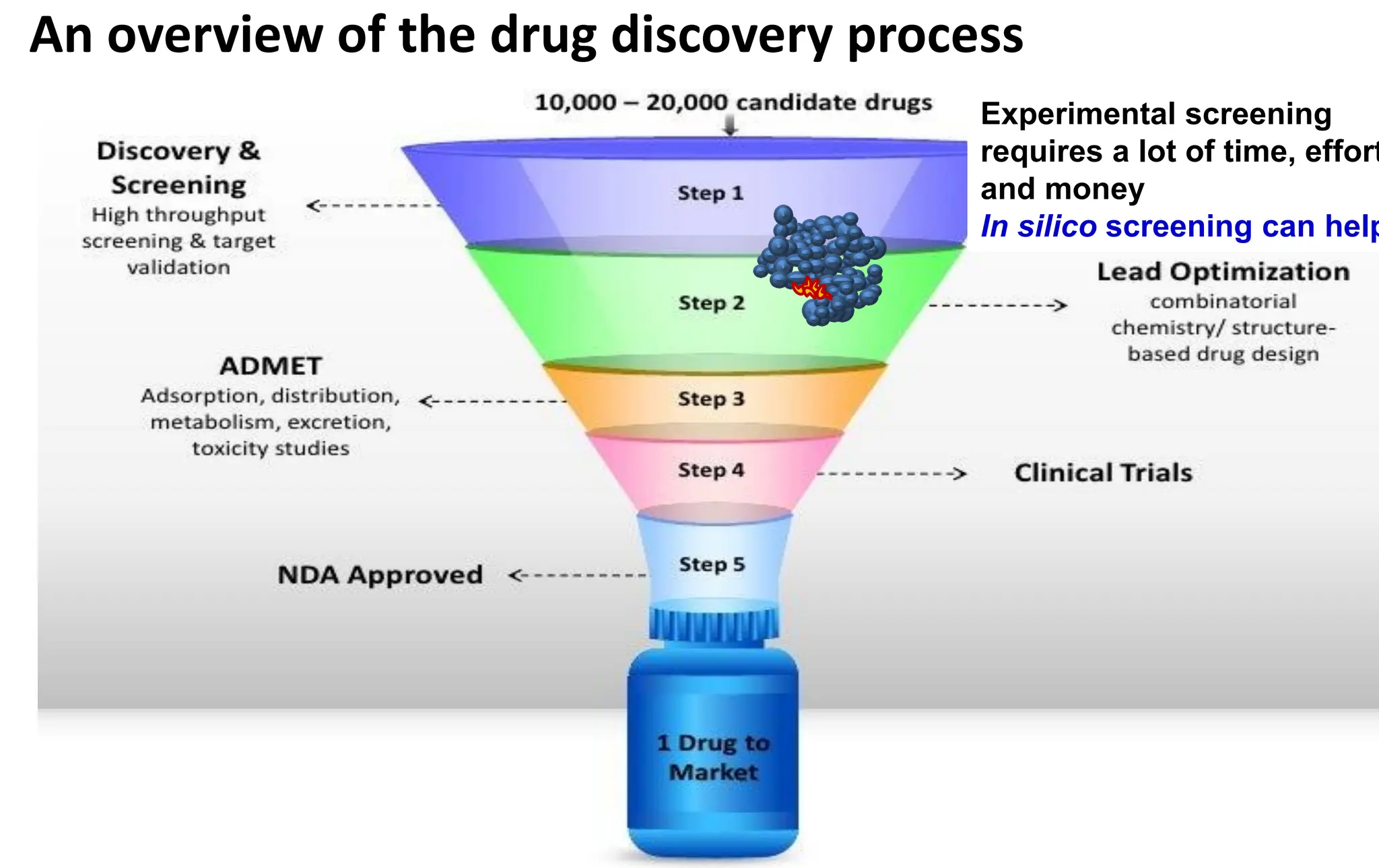
An overview ofthe drug discovery process
Experimental screening
requires a lot of time, effort
and money
In silico screening can help
4.
Pre-requisites for insilico screening
Structure of the target protein should be known
• Protein structure is the three-dimensional arrangement of atoms in an
amino acid-chain molecule.
• To understand the functions of proteins at a molecular level, it is often necessary to
determine their three-dimensional structure.
• Techniques such as X-ray crystallography, NMR spectroscopy, cryo electron microscopy
(cryo-EM) and dual polarisation interferometry to determine the structure of proteins.
• Protein structures range in size from tens to several thousand amino acids
• To be able to perform their biological function, proteins fold into one or more specific
spatial conformations driven by a number of non-covalent interactions such as hydrogen
bonding, ionic interactions, Van der Waals forces, and hydrophobic packing.
5.
Introduction to PDB
•Protein Data Bank- Databank for 3D structures of proteins,
nucleic acids, and complex assemblies.
• It contains
✓ 195,565 experimental Structures from the PDB archive
✓ 1,000,361 Computed Structure Models (CSM) from
AlphaFold DB and ModelArchive
• https://www.rcsb.org/ - demonstration on searching and
downloading the structures of proteins
6.
• Computational structureprediction methods that are used to determine
protein 3D structure from its amino acid sequence.
• Homology modeling predicts the 3D structure of a query protein
through the sequence alignment of template proteins.
• Homology modeling is one of the most accurate computational method
to create reliable structural models and is commonly used in many
biological applications.
• Generally,It involves four steps:
Target identification
Sequence alignment
Model building
Model refinement
What if the structure is not known for the
target?
7.
• Many toolsare available for
homology modelling.
Swiss model
Modeller
FoldX
Phyre and Phyre2
HHPred
ROBETTA
Homology search
(BLASTp)
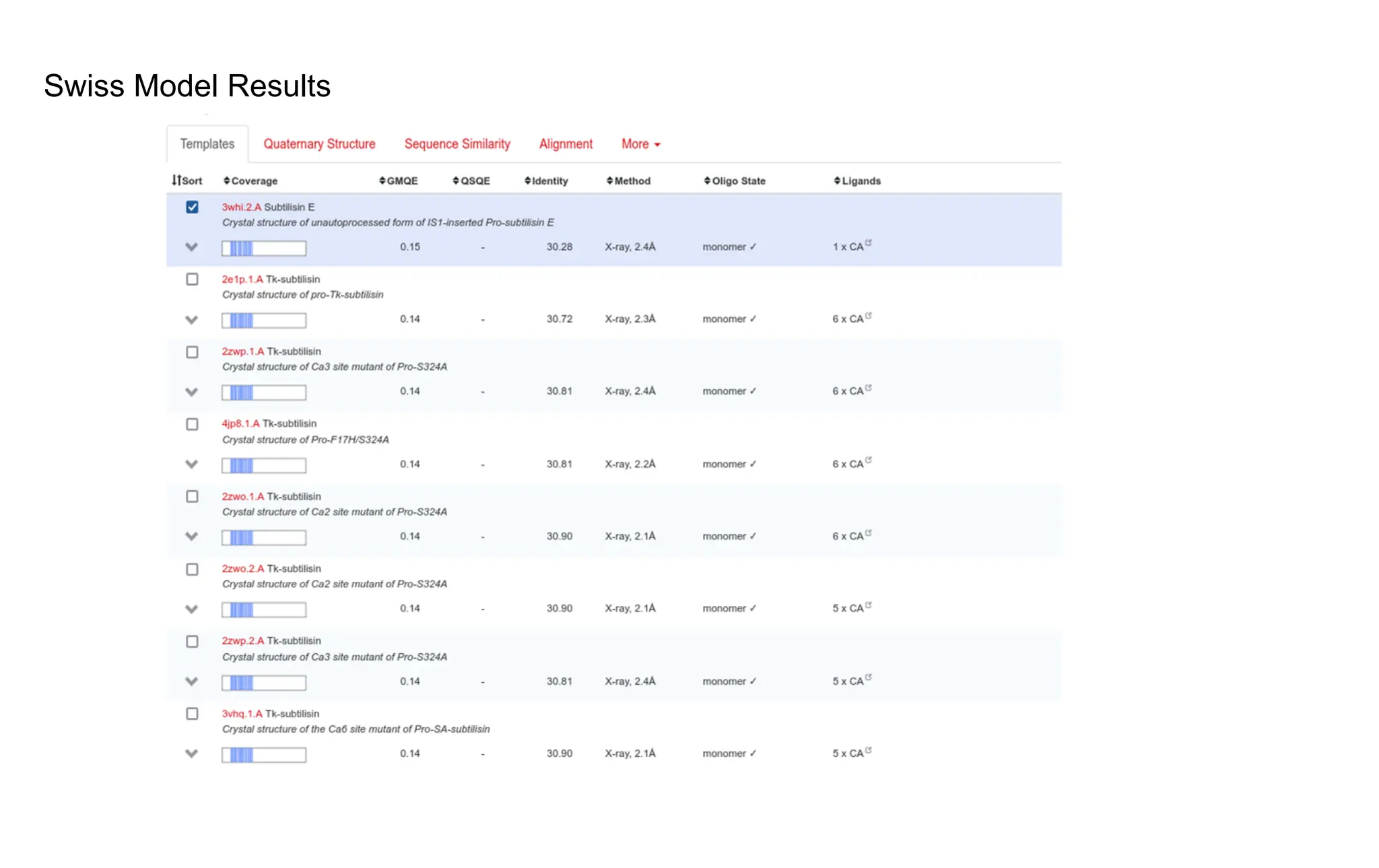
Swiss-Model
Select the model based on
best % similarity and QMEAN
Download predicted Structure
Homology modelling workflow
8.
SWISS- MODEL forHomology Modelling
• is a fully automated protein structure homology-modelling server
• accessible via the Expasy web server https://swissmodel.expasy.org/
or
• from the program DeepView (Swiss Pdb-Viewer)
• Demonstration on modelling using Swiss model server -
https://swissmodel.expasy.org/
The sequence similarityis less than 40% and the quality check
methods like ramachandran plot, global score show error in the
structure.
Solution-1:
we can try to model the domain part using the homology
modelling
Limitation: It shows less sequence similarity.
Solution-2:
The other way to model the structure with ab-initio method.
If any limitations from Homology modelling
11.
What are thedrugs?
• Small molecules that bind to a specific protein that is important to cure the disease
• Which drugs / molecules to choose → experiments but it’s expensive and
time consuming
• In silico analysis is cheaper and faster to find which small molecule can bind well
to the protein
• There are many chemical compound databases available
• PubChem - https://pubchem.ncbi.nlm.nih.gov/
• ZINC - https://zinc.docking.org/
• IBS (Inter Bioscreen) Database -
http://mastersearch.chemexper.com/misc/hosted/ibscreen/
• ChemStar
12.
Molecular Docking
• Moleculardocking - most widely utilized computational phenomenon in the field of
computer-aided drug design (CADD).
• It is being utilized at the academic level as well as in pharmaceutical companies
for the lead discovery process.
• Molecular docking is a low-cost, safe, and simple-to-use technique that aids in
the investigation, interpretation, explanation, and discovery of molecular features
through the use of three-dimensional structures.
• Docking is a mathematical technique used to anticipate the structural interactions
of two or more chemical molecules.
• Is mainly associated with two terms: ligand and protein.
• Protein is the target site where ligand may bind to give specific activity.
• Molecular docking provides information on the ability of the ligand to bind with
protein which is known as binding affinity.
13.
Molecular Docking
• Usingscoring functions, it is possible to estimate the strength of the connection or
binding affinity across two compounds based on their preferential orientation.
• There are two distinct forms of docking.
• Rigid docking
• Flexible docking
• Rigid docking - the compounds are inflexible, we are seeking a rearrangement
of one of the compounds in three-dimensional space that results in the best
match to the other compounds in parameters of a scoring system
• Flexible docking – the compounds are in flexible to identify the better
conformation
14.
Molecular Docking
• Modelsof Molecular Docking
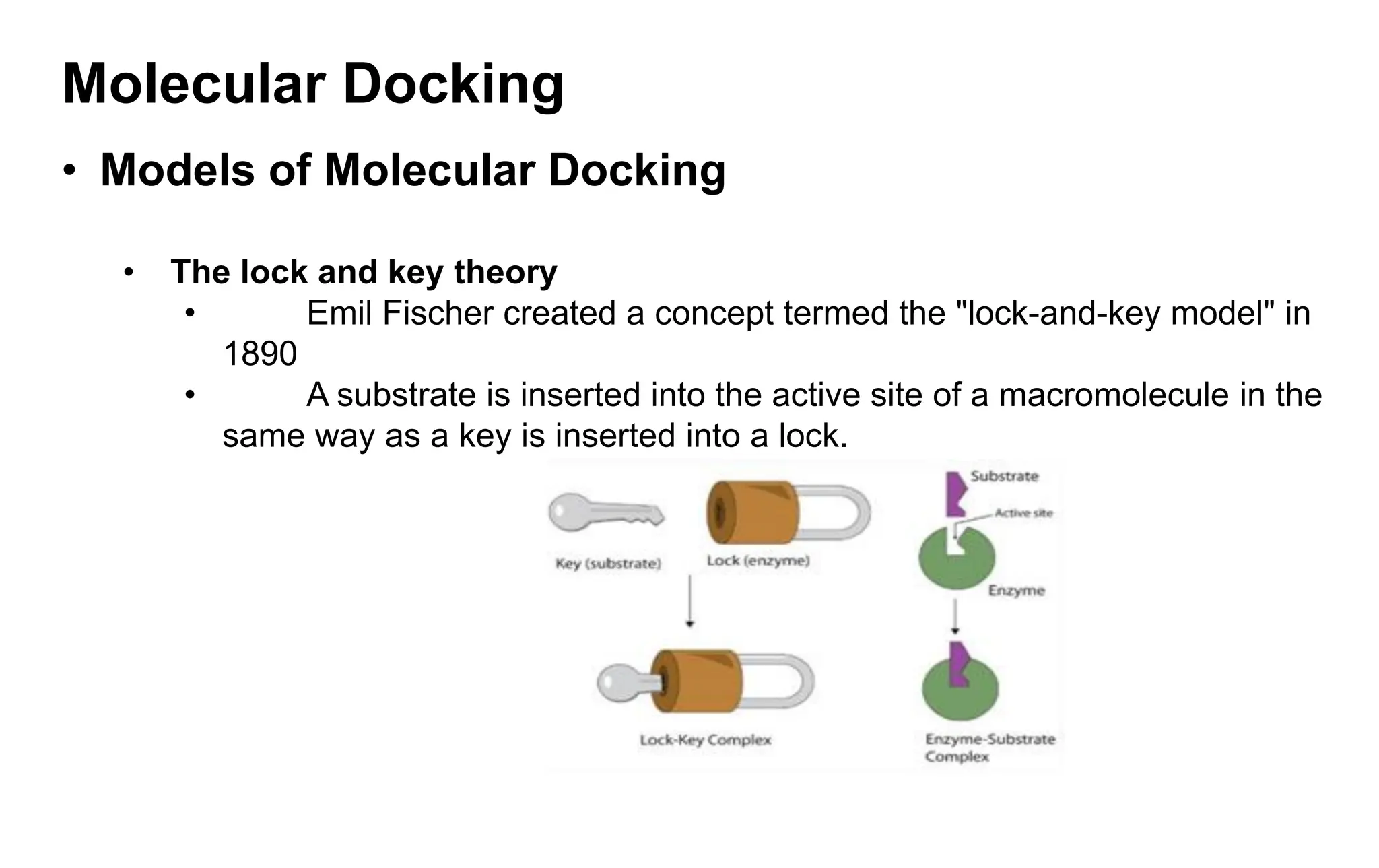
• The lock and key theory
• Emil Fischer created a concept termed the "lock-and-key model" in
1890
• A substrate is inserted into the active site of a macromolecule in the
same way as a key is inserted into a lock.
15.
Molecular Docking
• Modelsof Molecular Docking
• The induced-fit theory
• Daniel Koshland proposed the "induced fit theory" in 1958
• The fundamental concept is that throughout the character
recognition, both the ligand and target.
16.
Molecular Docking
• Modelsof Molecular Docking
• The conformation ensemble model
• Proteins have been discovered to undergo significantly greater
conformational changes.
• According to a new concept, proteins are composed of a pre-
existing ensemble of conformational states.
• The protein's flexibility enables it to transition between states
17.
Molecular Docking
• Approachesof Molecular Docking
• Monte carlo approach
• It creates a randomized conformation, translations, and rotation of a
ligand in an active site. It assigns an initial configuration value.
• Then it develops and scores a new configuration. It determines if the new
configuration is kept using the Metropolis criterion.
• Matching approach
• The optimal location of the ligand atom in the site is determined, resulting
in a ligand-receptor arrangement
18.
Molecular Docking
• Approachesof Molecular Docking
• Ligand fit approach
• Quick and precise methodology for docking small molecules ligands into
protein active sites while taking shape complementarity into account
• Point complimentarily approach
• These techniques are focused on comparing the shapes and/or chemical
properties of different molecules.
• Blind docking - identifies the potential ligand binding sites
• Fragment-based method
• Dissolving the ligand into individual photons or particles, attaching the
fragments, and lastly connecting the fragments
19.
Molecular Docking
• Approachesof Molecular Docking
• Distance geometry
• The distance geometry framework enables the assembly of these inter-
and intra- molecular distances and the calculation of three-dimensional
structures that are compatible with them
• Inverse docking
• Docking a specific small molecule of interest to a library of receptor
structures.
• The technique can be used to identify new potential biological targets of
known compounds
20.
Molecular Docking
• Requirementsfor Molecular Docking
• A ligand docking strategy involves the following elements:
✓ A target protein design
✓ The compounds of interest or a database comprising existent or
virtual compounds for the docking process
✓ A computational foundation that enables the appropriate docking
and scoring methods to be implemented
• The majority of docking algorithms consider the protein to be stiff, whereas
the ligand is often considered to be flexible.
• Apart from the structural degree of freedom, the bonding position of the
protein in its binding pocket must be considered.
21.
Molecular Docking
• Applicationsof Molecular Docking
• Hit identification in drug discovery
• It enables rapid screening of vast databases of possible medications in
silico to find compounds that are capable of binding to a particular target
of interest
• Lead optimization in drug discovery
• It’s used to anticipate the location and relative position of a ligand's
interaction to a protein
• Remediation in which docking can be utilized to forecast which contaminants
are degradable by enzymes
22.
Molecular Docking
• Softwareavailable for Molecular Docking
• Autodock - Autodock-vina is one of the docking engines of the AutoDock
Suite.
Consists of a three-dimensional lattice of regularly spaced points encircling
and centered about the macromolecule's region of interest.
• Flex-X - Using the "position clustering" technique, the base fragment is picked up
and docked.
• Gold - Genetic Enhancement and Receptor Docking
• ArgusLab
• Hammerhead
• ICM
• MCDock
• GemDock
• Glide
• Yucca
23.
Autodock –Vina
• Pre-requisitetools/softwares for running AutoDock-vina
• MGLTools
• OpenBabel/Pymol for conversion
• Autodock-vina
• Pymol for visualization
• Required files for docking with AutoDock Vina:
• Pdbqt files of protein and the ligand
• Configuration file
• Grid file
24.
• Steps inAutoDock Vina
1. Preparation of PDB file before docking
• Download a protein crystal structure from PDB
We are using Human Serum Albumin complexed with 3-carboxy-4-
methyl-5-propyl-furanpropanoic acid (CMPF) (PDB ID: 2BXA).
• Open the PDB file and remove HETATOMS - HETATM in PDB
• After removing hetatoms, we will keep only one of the twochains (here, Chain
A was taken) and remove the rest of the three chains and save this file as
“protein.pdb”
• Now save the file as “protein.pdb”.
25.
• Steps inAutoDock Vina
2) Preparation of ligand before docking
• Open PubChem (www.pubchem.ncbi.nlm.nih.gov) and search for the compound.
We are using “sodium octanoate” as a ligand.
• Click on Sodium octanoate and look under “3D Structure” section, click on “Download”
and then you will see four different formats for downloading it.
• We will download the .SDF format.
• Since we need the protein and the ligand to be in a .pdb format, therefore, we have to
convert .SDF to .pdb. We will use PyMol for this purpose
• Open PyMol, and open the downloaded ligand. Click on “File” –> “Export Molecule” –
> select the molecule –> click “OK”. You can save it to your desired folder.
26.
3. Preparation of.pdbqt files
Ligand
• First, we will prepare a .pdbqt file of the ligand. PDBQT - Protein Data Bank, Partial
Charge (Q), & Atom Type (T)) format.
• Open AutoDock Vina –> click “Ligand” –> click “Input”–> click “Open”
• It will ask to select your ligand, we will go to the folder where we have saved our
ligand’s .pdb file and click “SO.pdb”.
• Click “Ligand” –> click “Torsion Tree” –> click “Detect Root”. It will show the torsion
angle on the ligand from where it can be rotated.
• Click “Ligand” –> click “Output” –> Click “Save as PDBQT”.
• We can rename the ligand, but we will use the same name as before and will name it as
“SO.pdbqt” and save it in the same folder.
27.
Receptor
• Open AutoDockVina, click “File” –> click “Read Molecule” –> select protein.pdb.
• We will delete water molecules from the protein as they can make unnecessary bonds
with the ligand. Click “Edit” –> “Delete water”.
• We will add polar hydrogens in order to avoid any empty group/ atom left in the
protein. Click “Edit” –> click “Add Hydrogens” –> click “Polar only”.
• We will save this file as .pdbqt, click “Grid” –> click “Macromolecule” –> click “Choose”
–> select the “protein.pdb” –> click “OK”. It will ask for a folder to save, then save it as
“protein.pdbqt”, in the same folder where the pdbqt file of the ligand was saved.
28.
4. Defining bindingsite
• Now we will define the binding site in the protein
• For example, in this protein, we will look for Tyr150, Lys199, Arg222, Arg257, and His242
because this is the binding site of CMPF in human serum albumin and we want to bind
another ligand in the same position.
5. Defining Grid Box for docking
Click “Grid” –> click “Grid Box”.
Now try to adjust the grid box by scrolling the three coordinates, such that it covers all the
selected residues.
After adjusting the grid box, click “File” –> click “Output Grid Dimension File” –> save this
file as grid.txt in the same folder.
Click “File” –> Click “Close saving current”.
29.
6. Preparation ofConfiguration file
AutoDock Vina requires an input configuration file that contains all the information of the
parameters used in configuring the docking including the name of the protein and the
ligand.
7. Perform Docking
Put all the following in the same folder (i.e., dock):
• 2bxa.pdbqt
• SO.pdbqt
• conf.txt
• All the MGL_Tools, Autodock Tools, Python.exe (for Linux) and Autodock Vina
setup files.
30.
7. Perform Docking
Putall the following in the same folder (i.e., dock):
• protein.pdbqt
• SO.pdbqt
• conf.txt
• All the MGL_Tools, Autodock Tools, Python.exe (for Linux) and Autodock Vina
setup files.
Linux
Open the terminal and enter into the “dock” folder.
Type the following command:
vina --config conf.txt Press “enter”.
Windows
Open the command prompt and enter the folder where all the docking files are
placed.
Type the following command:
vina.exe -–config conf.txt -–log logSO.txt
31.
Vina Output
After thesuccessful docking, you will get a log file, which in this case is named “logSO.txt”.
The log file will be as follows:
• This file consists of all the poses
generated by the AutoDock Vina along
with their binding affinities and RMSD
scores.
• In the Vina output log file, the first pose
is considered as the best because it
has more binding affinity than the other
poses and without any RMSD value,
but you can choose the appropriate
pose and visualize it in PyMol viewer.
32.
Vina output analysisusing PyMol
• Open PyMol then go to 'File' --> 'Open' --> then
select the PDB file of your protein
• Now open the vina output file. Again go to 'File' -->
'Open' --> then select the pdbqt file
• Go to the tab 'all' --> click 'A' (it will show you a
drop-down menu) --> 'preset' --> 'ligand sites' -->
'cartoon'.
• You can also measure the bond lengths following
these steps:
• Go to 'Wizard' --> 'Measurement'.
• The best pose is supposed to be the first docking
pose generated by Vina which shows ‘zero’ RMSD
value and best binding affinity.