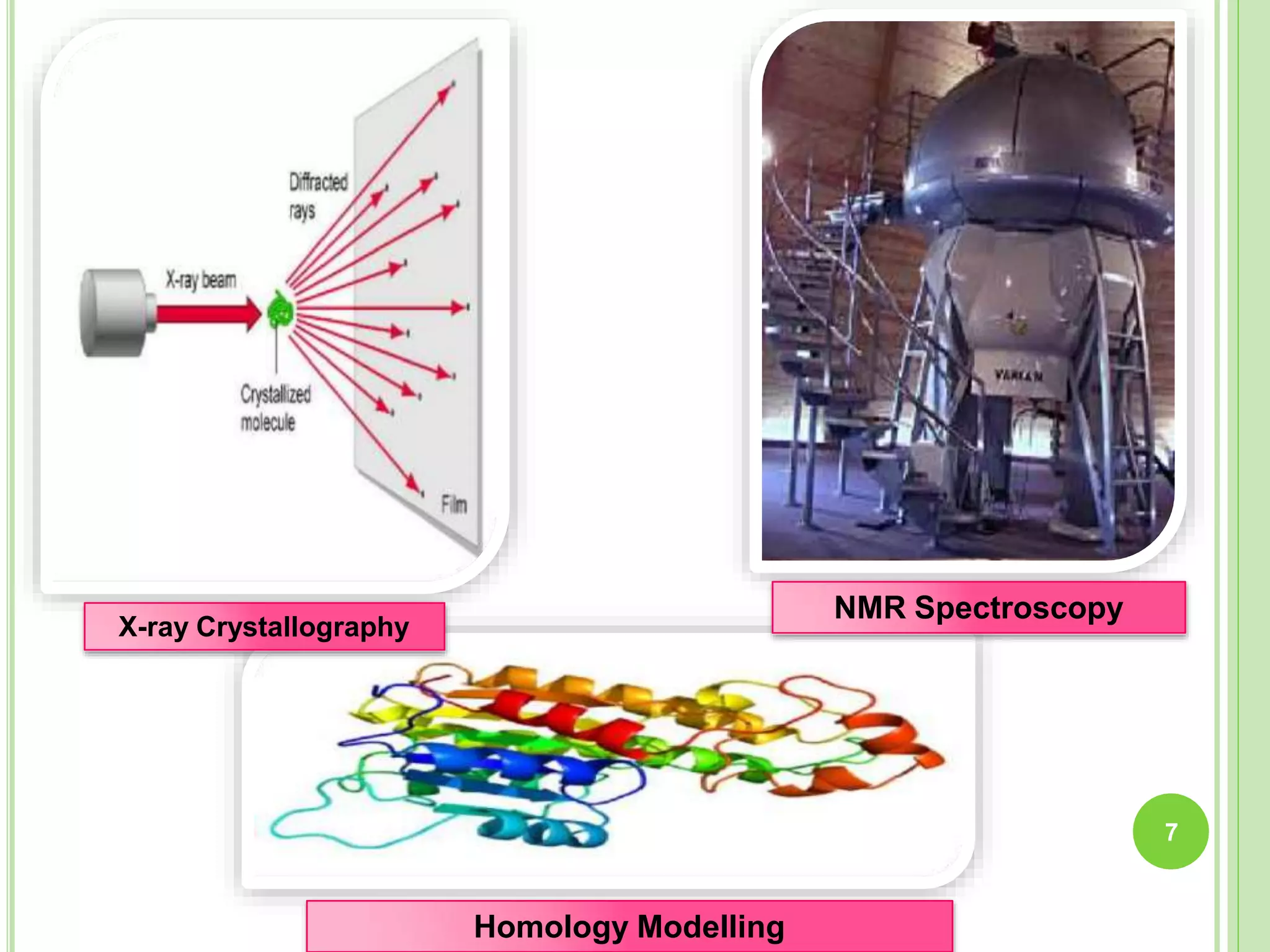
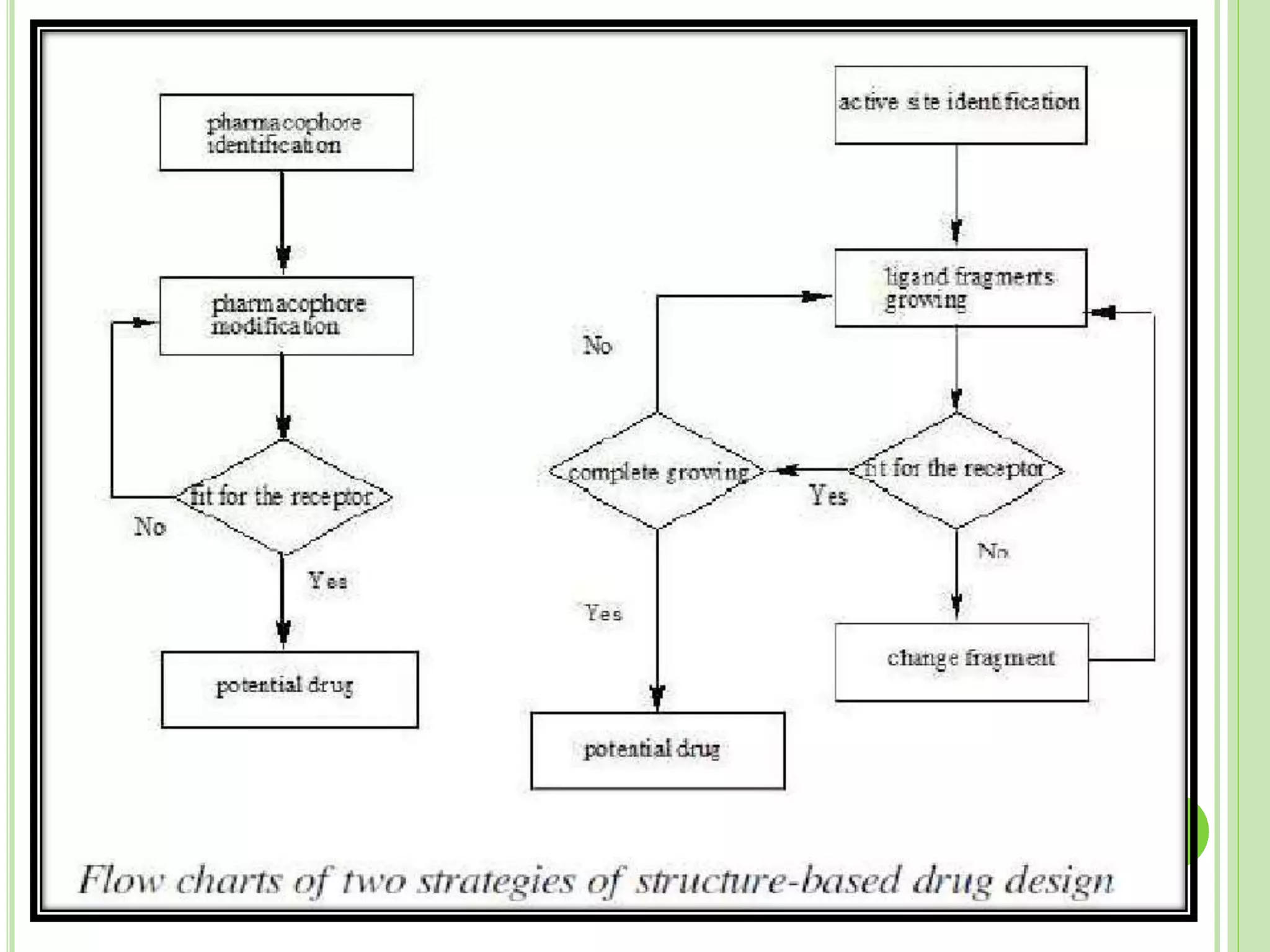
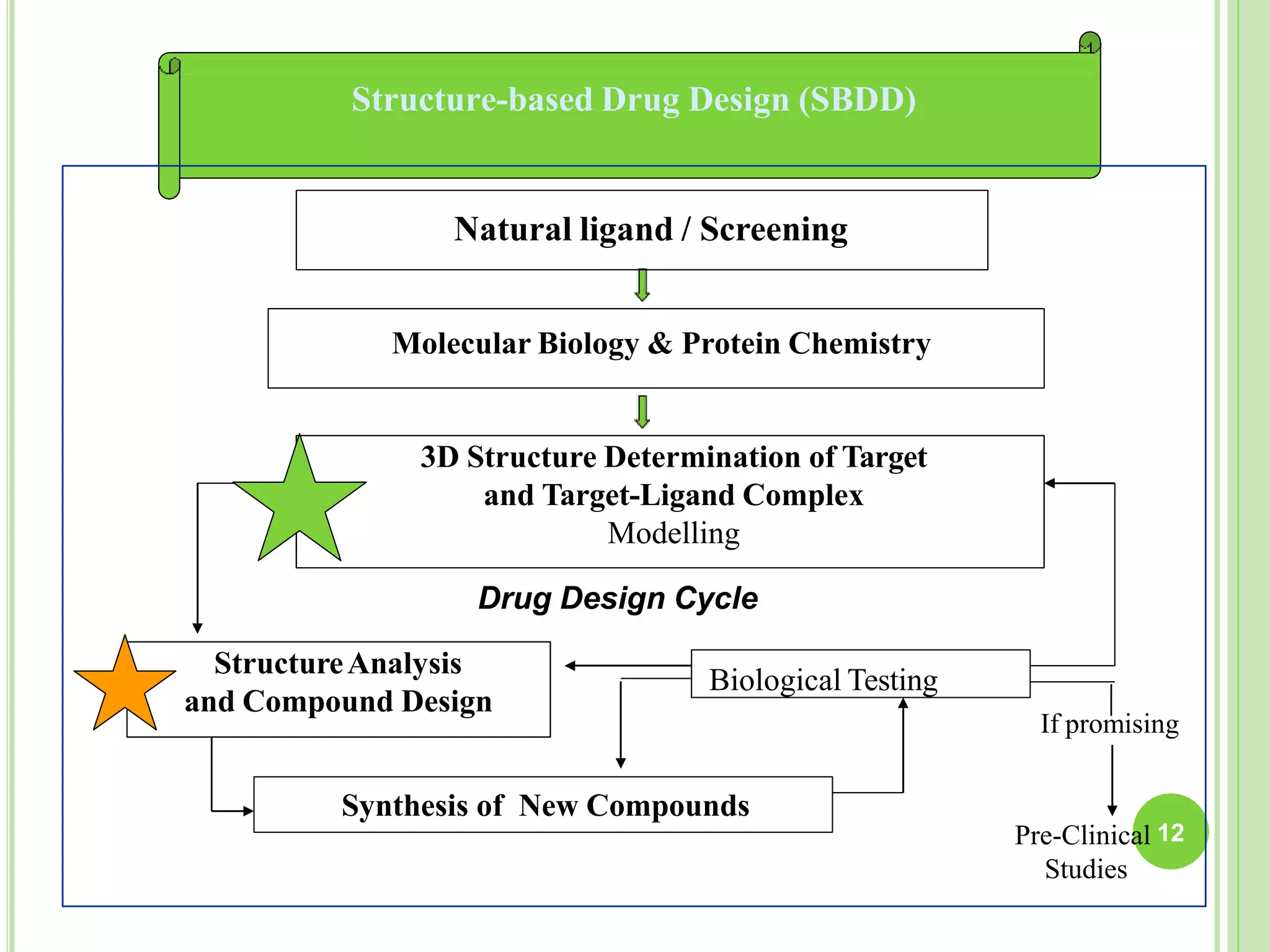
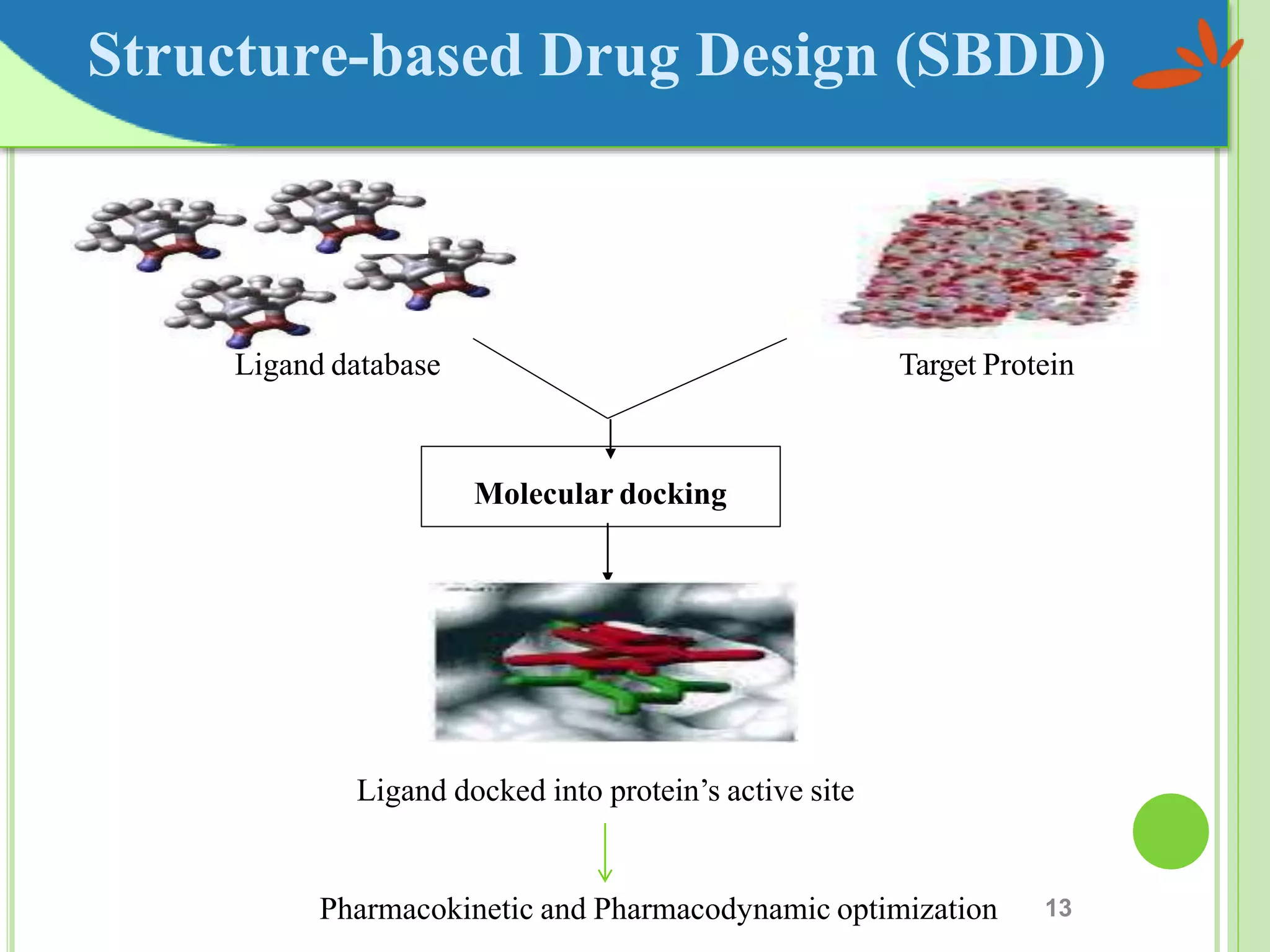
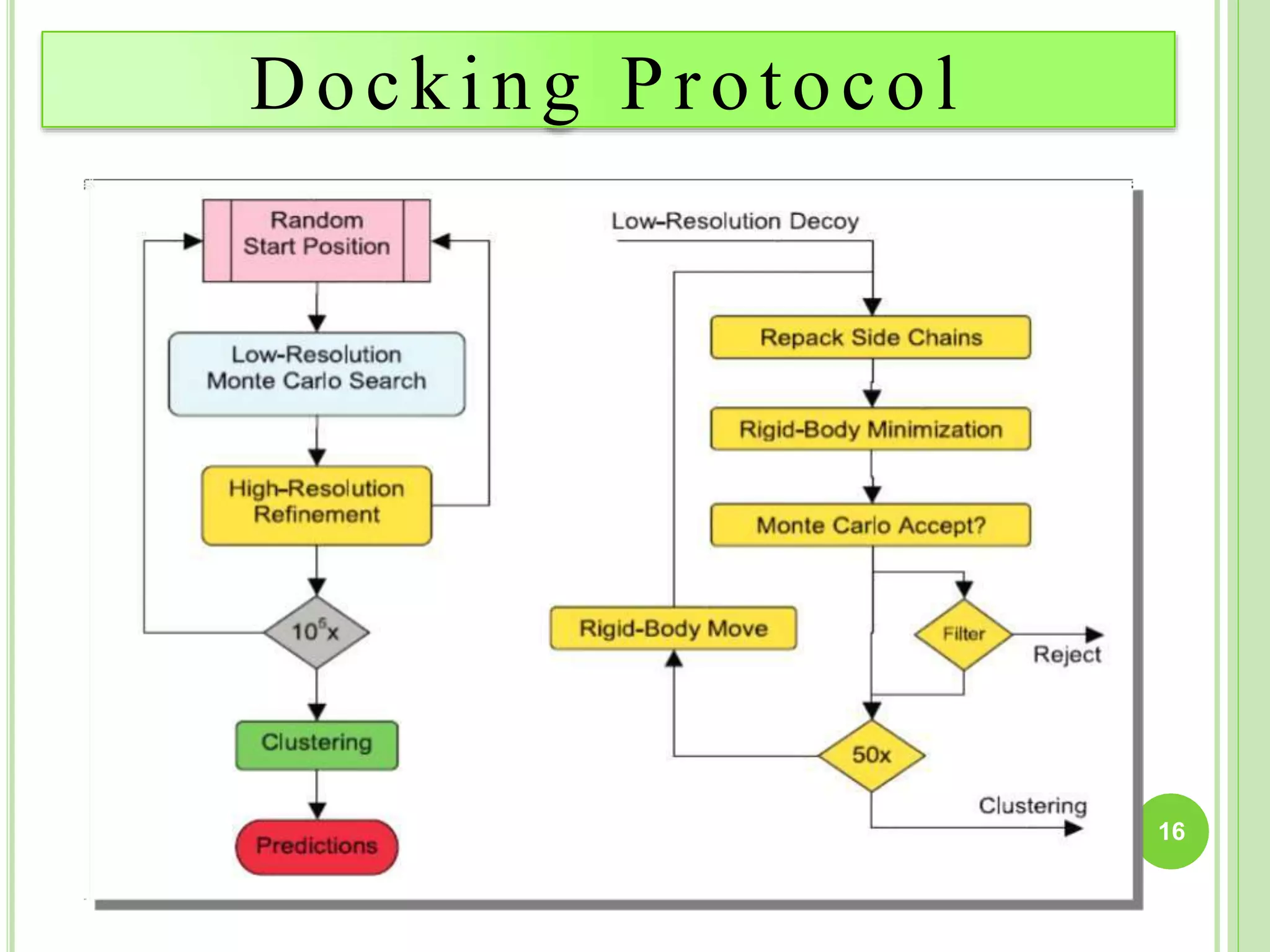
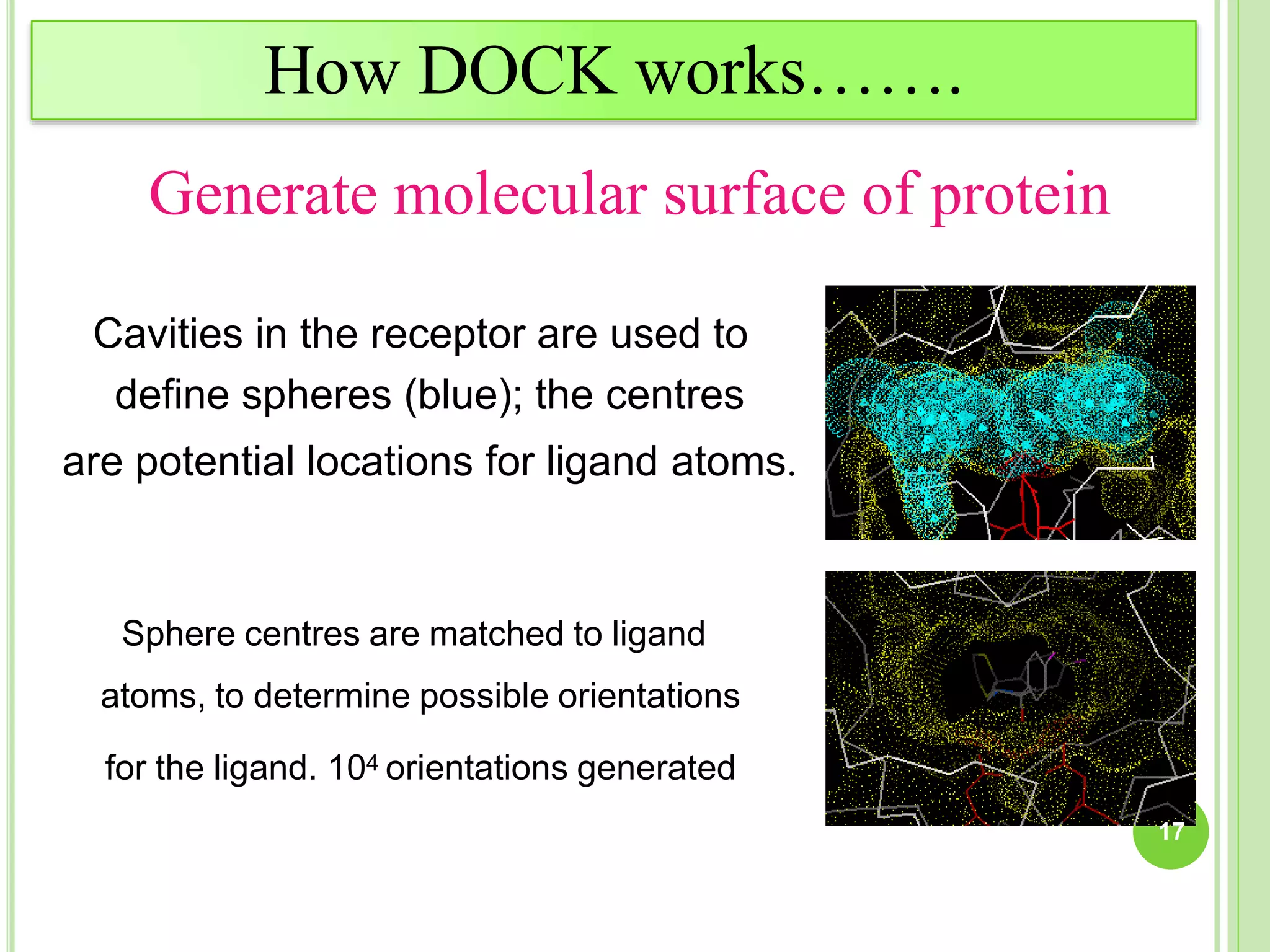
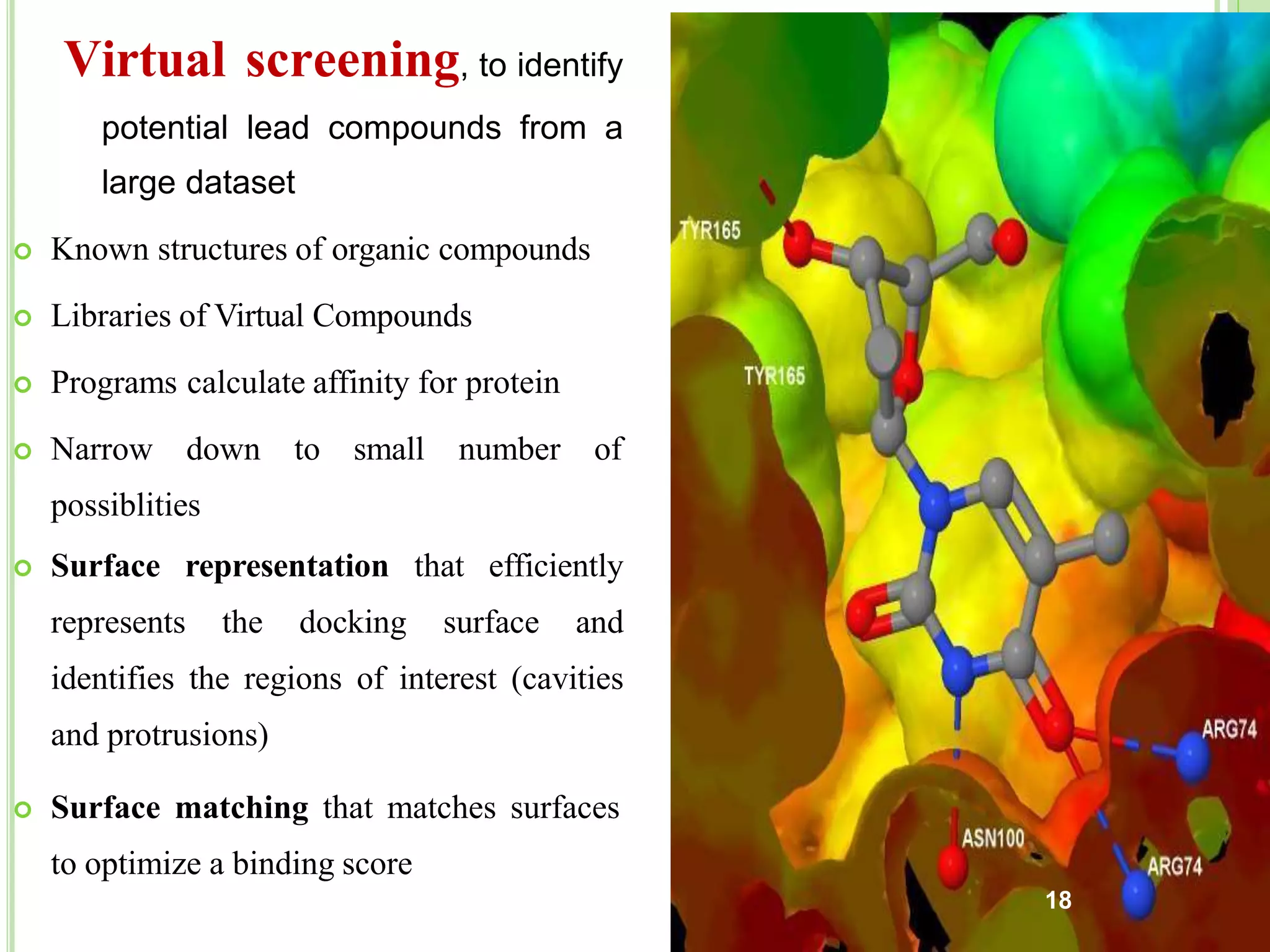
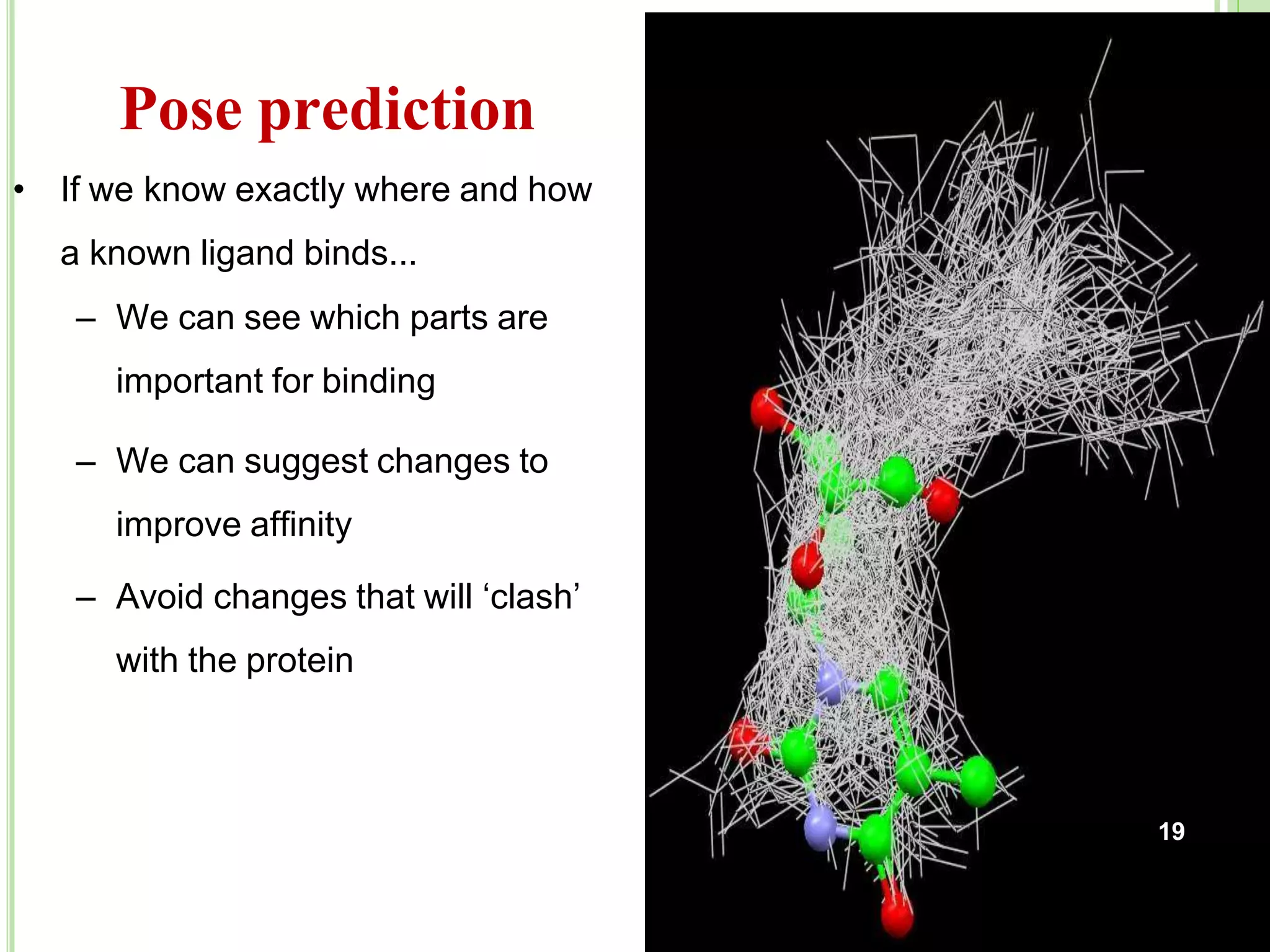
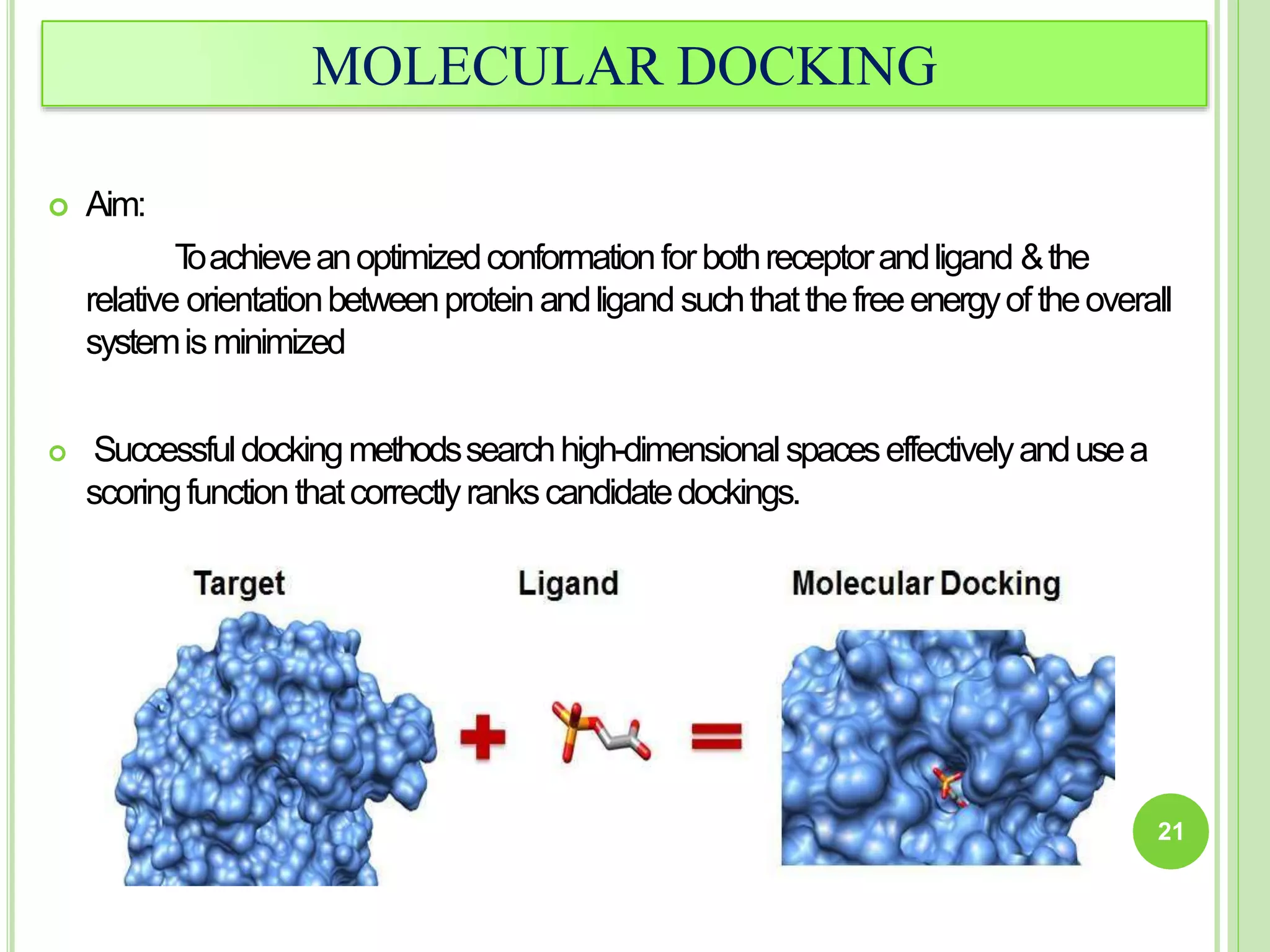
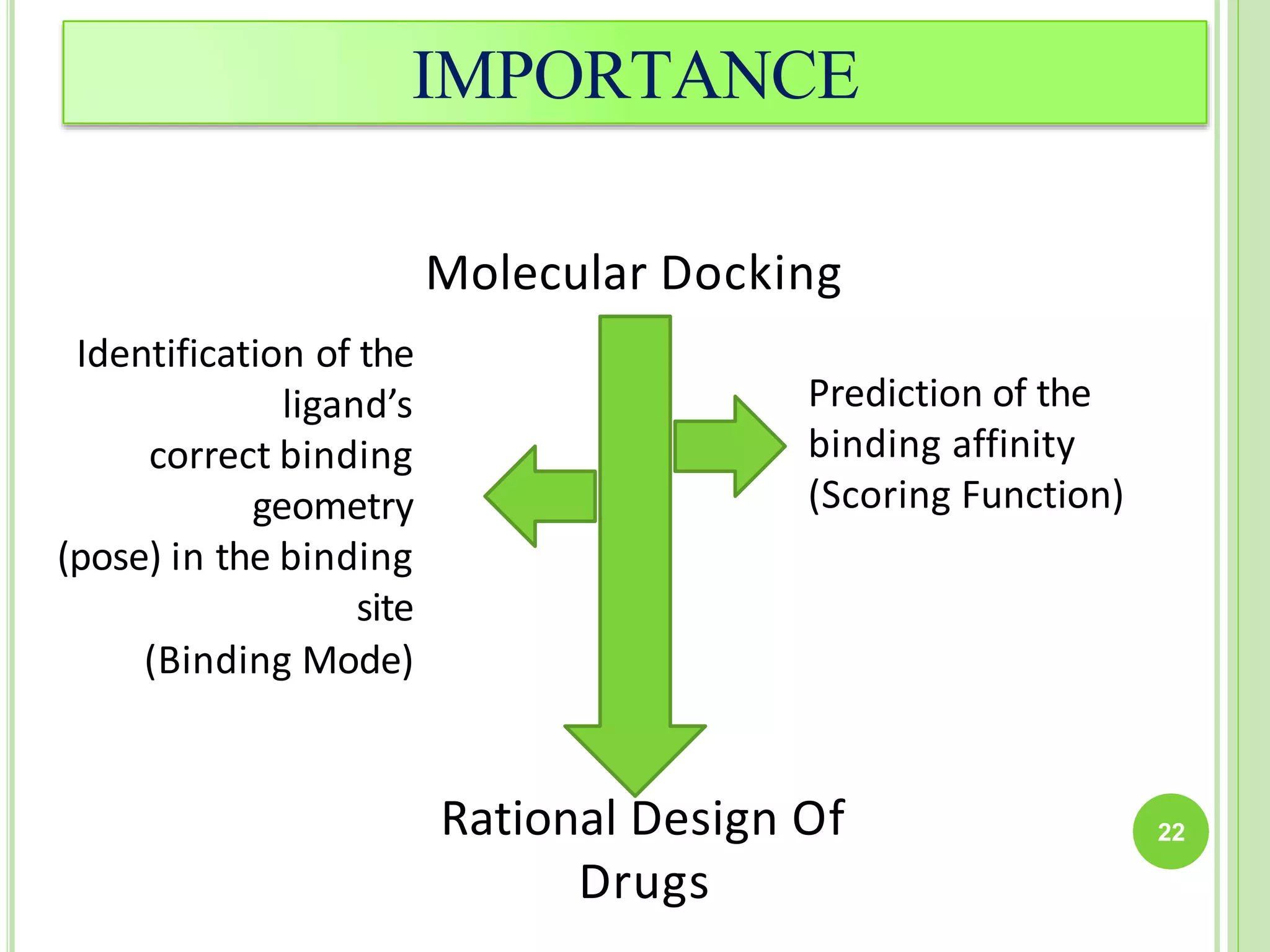
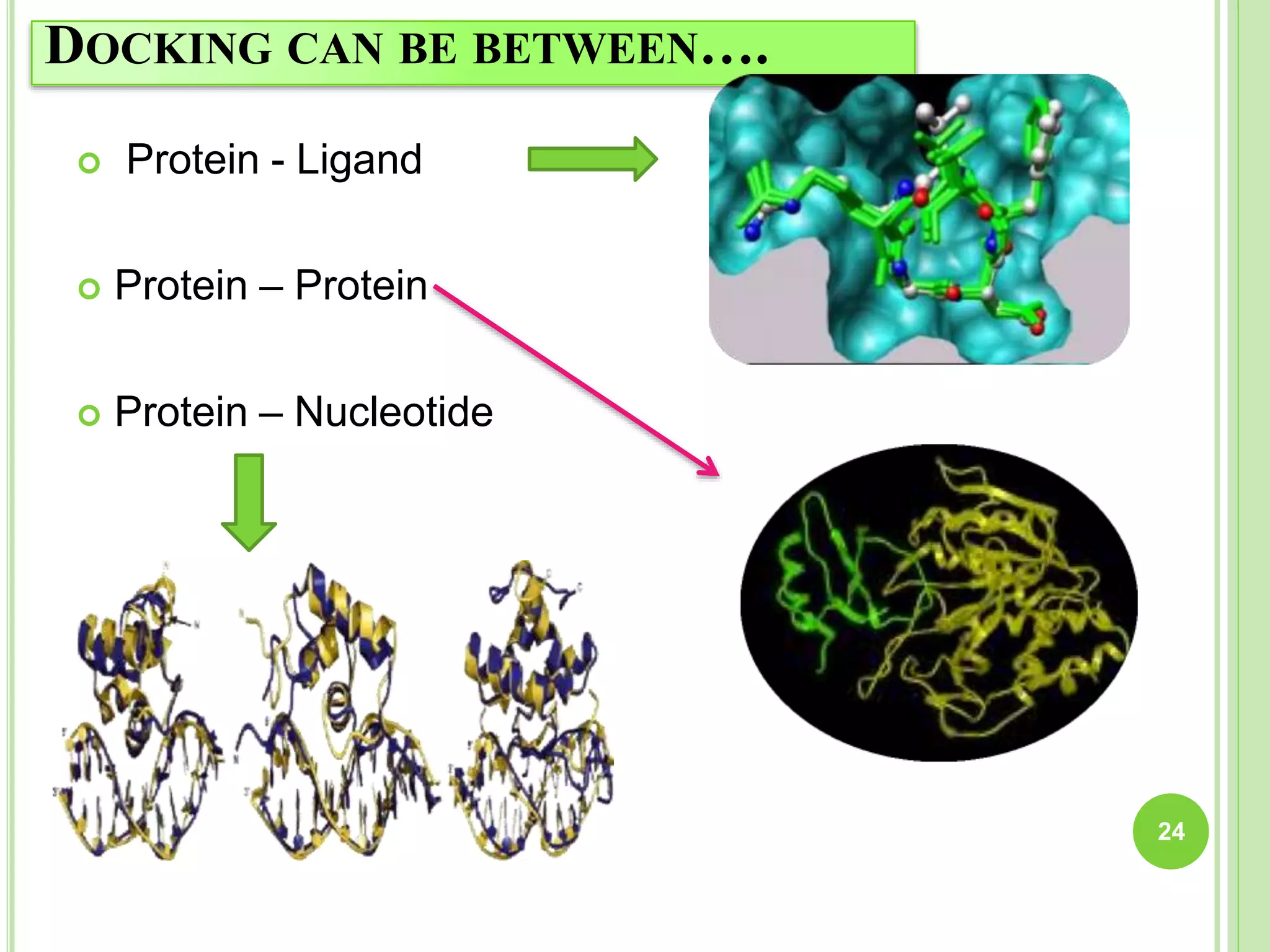
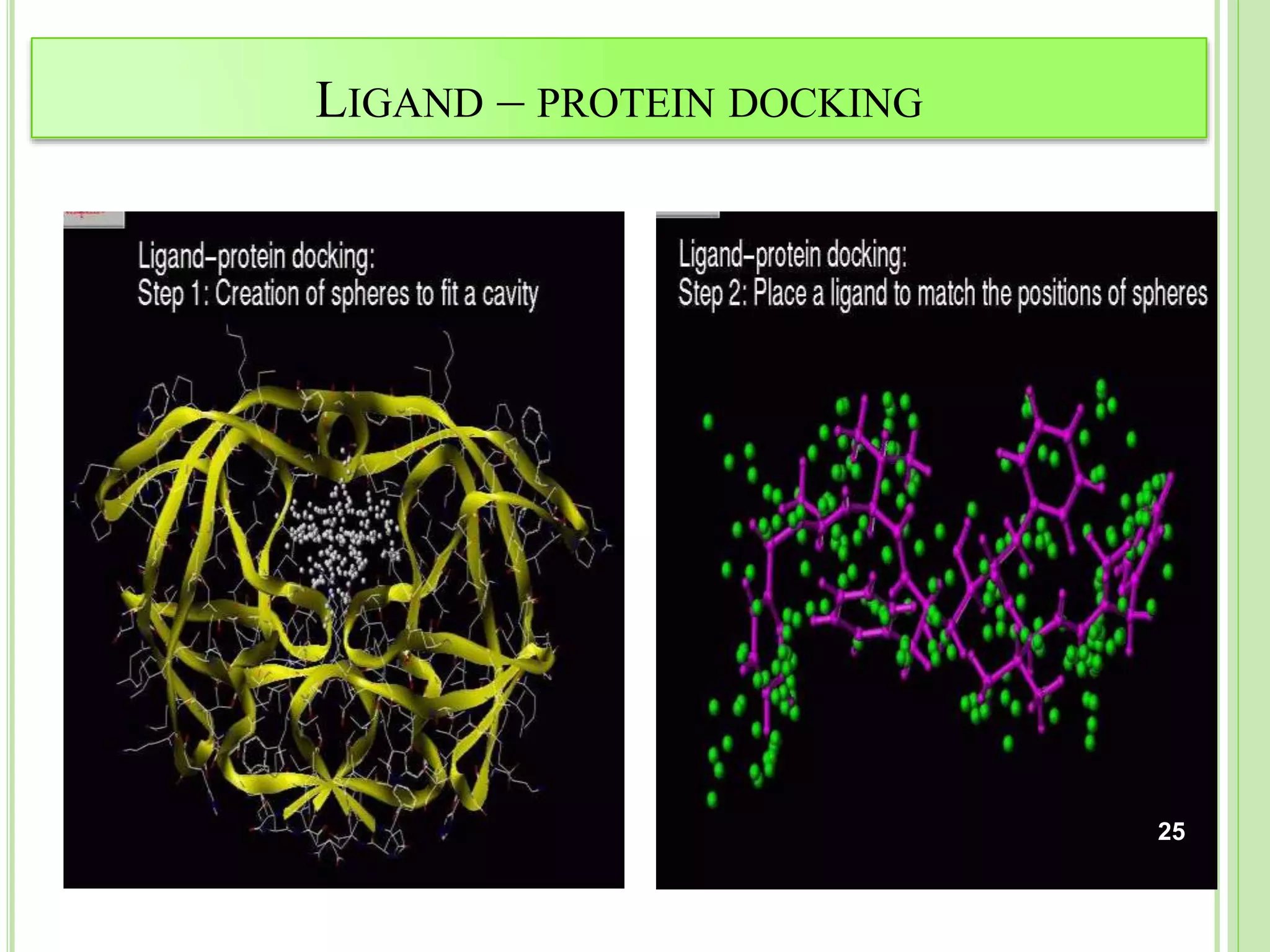
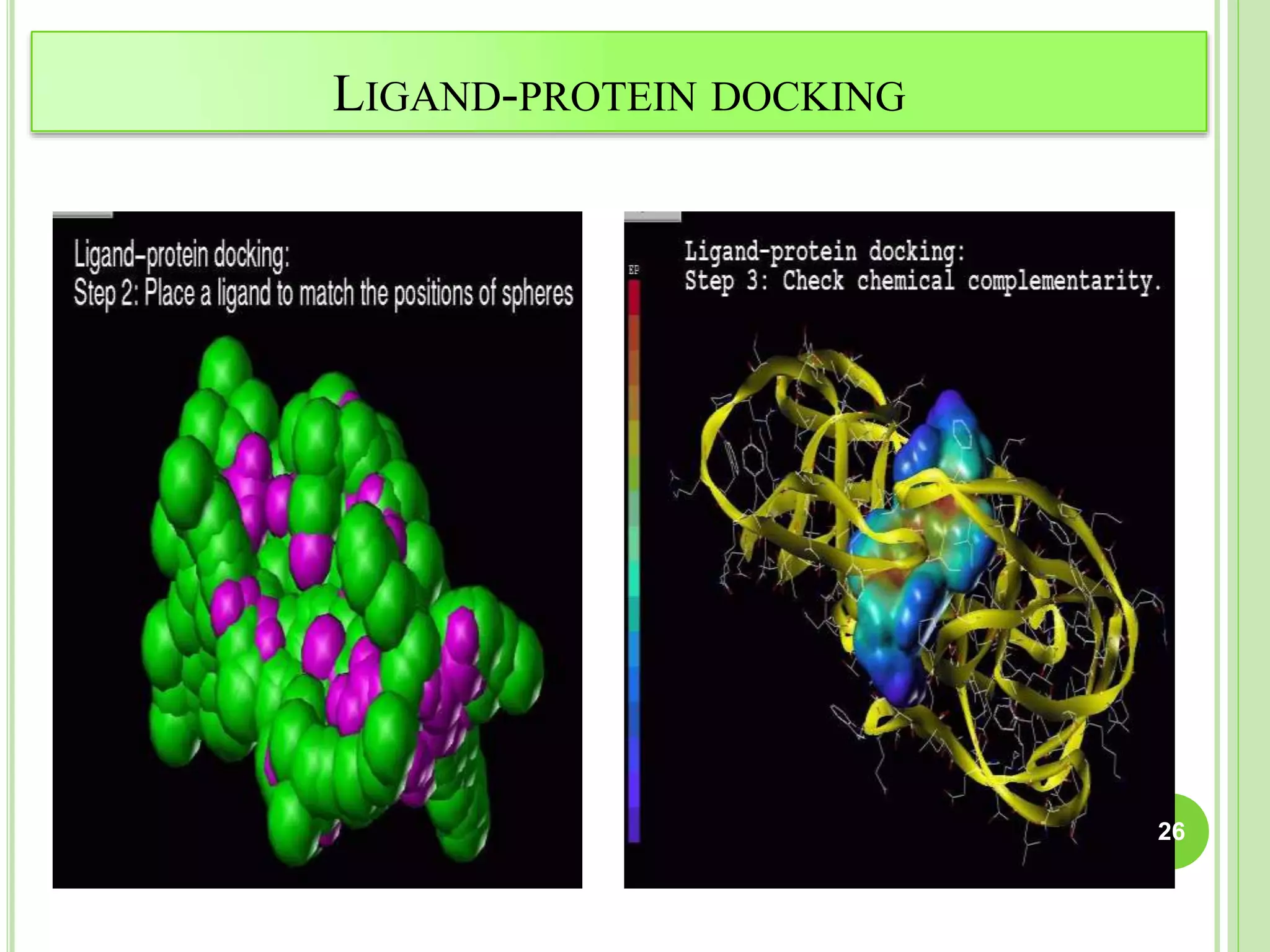
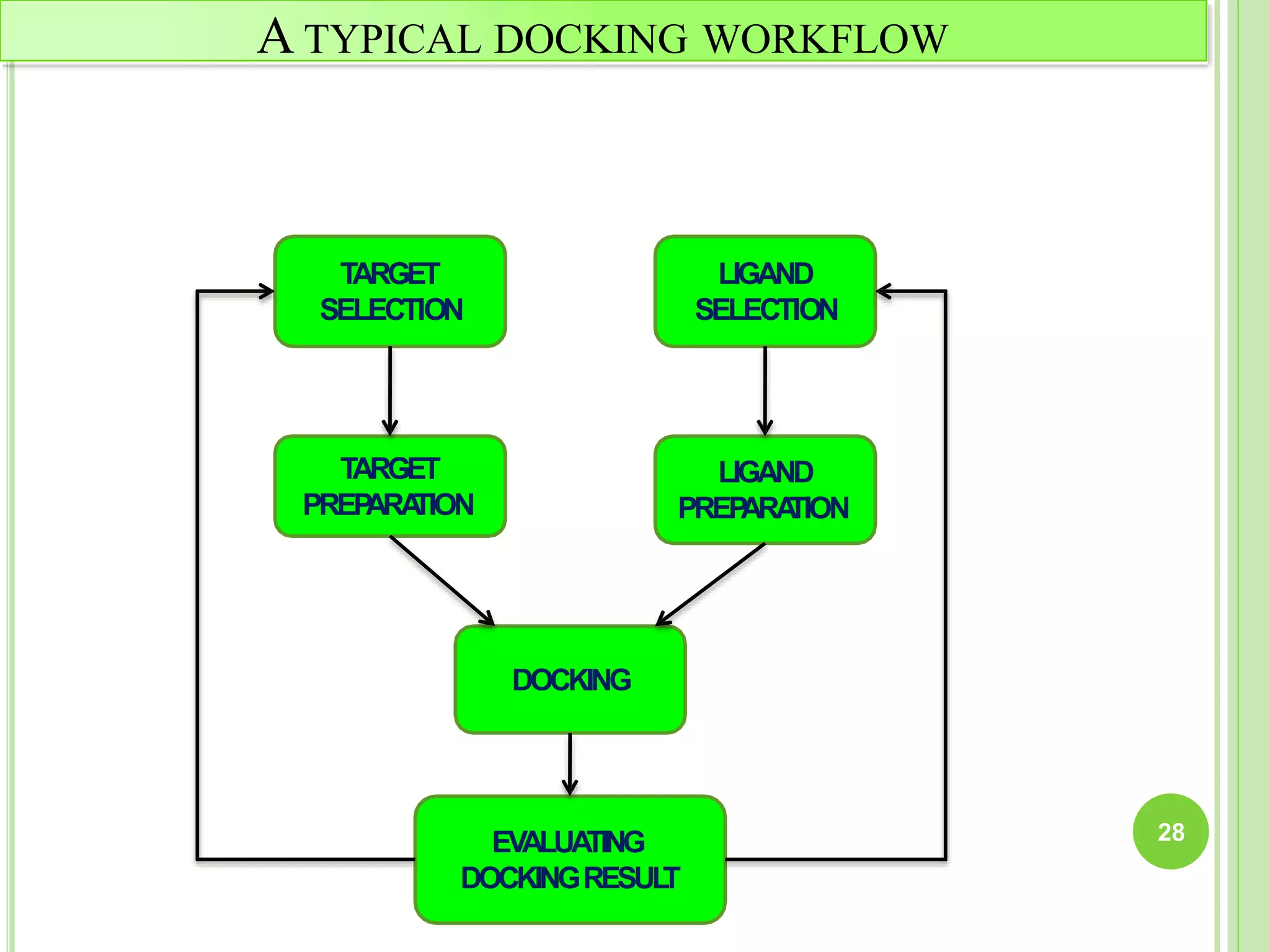
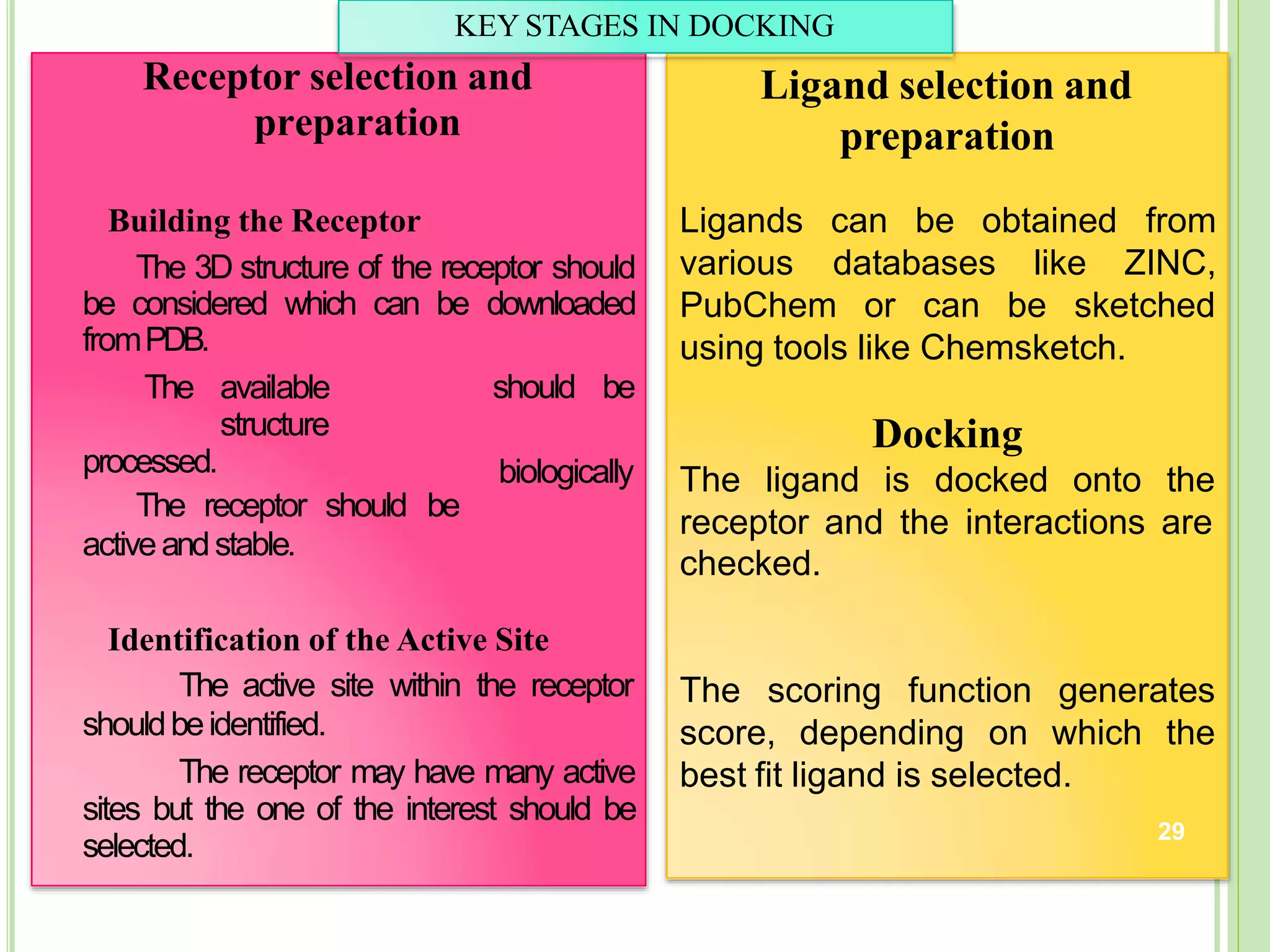
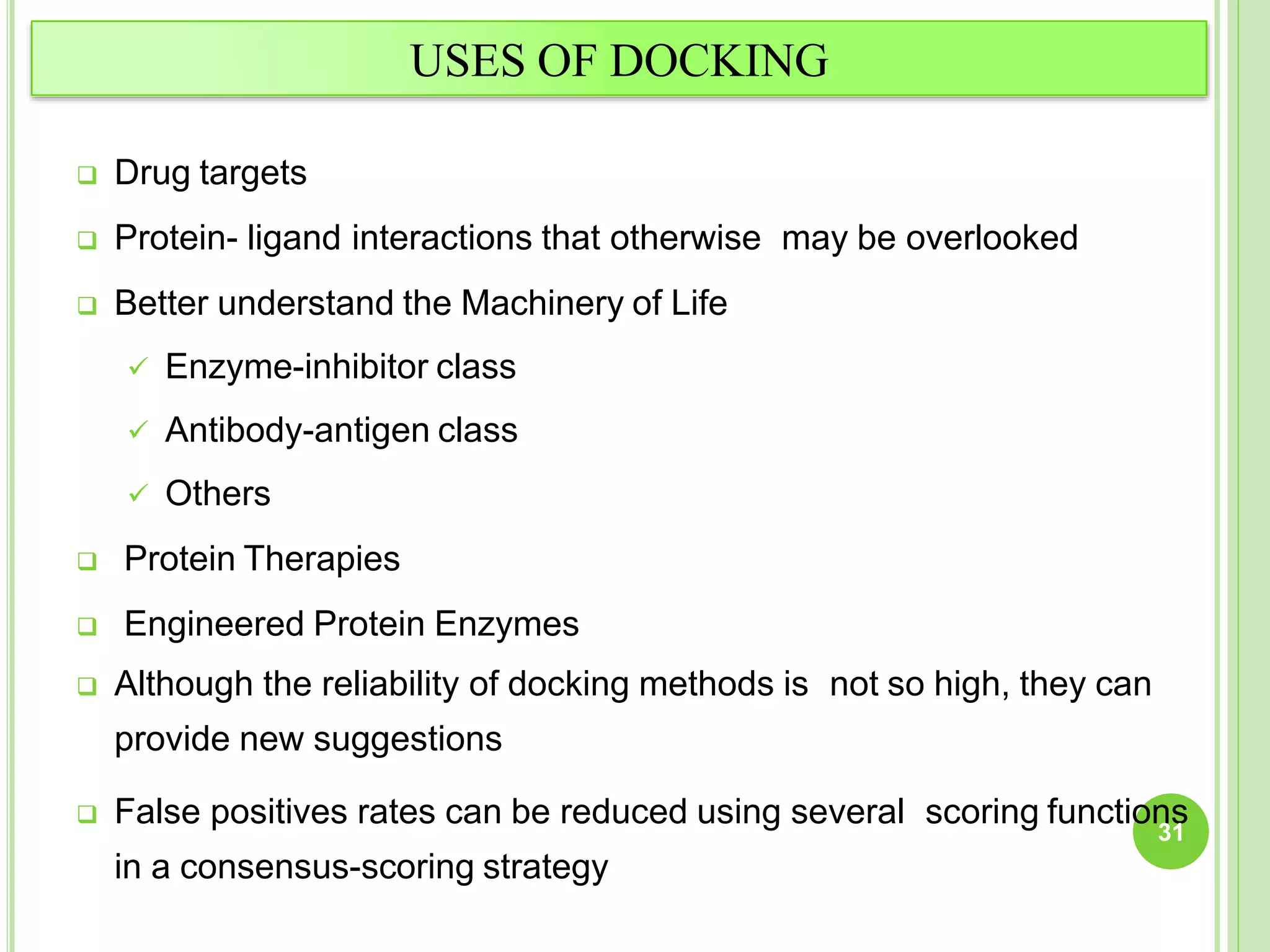
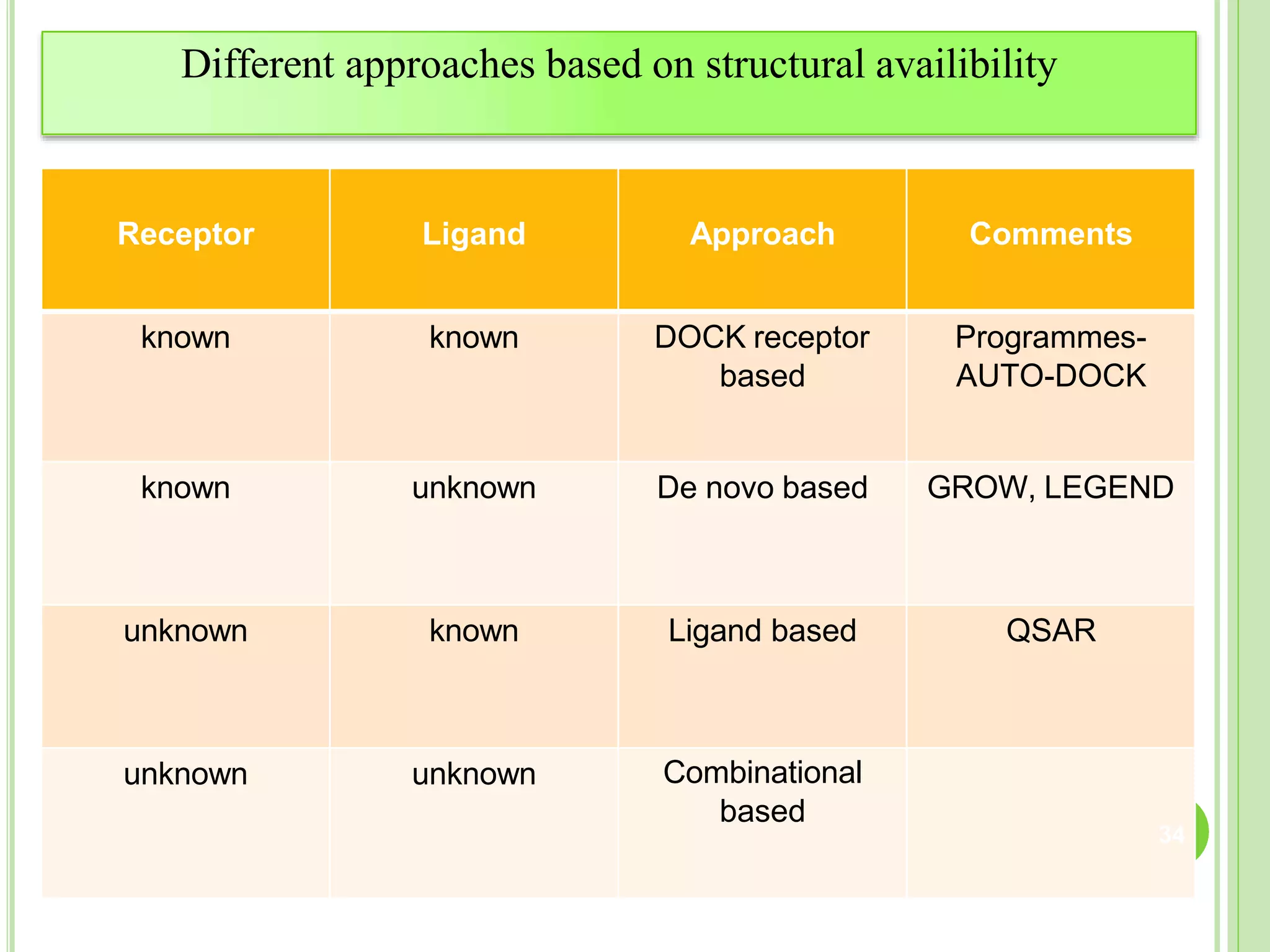
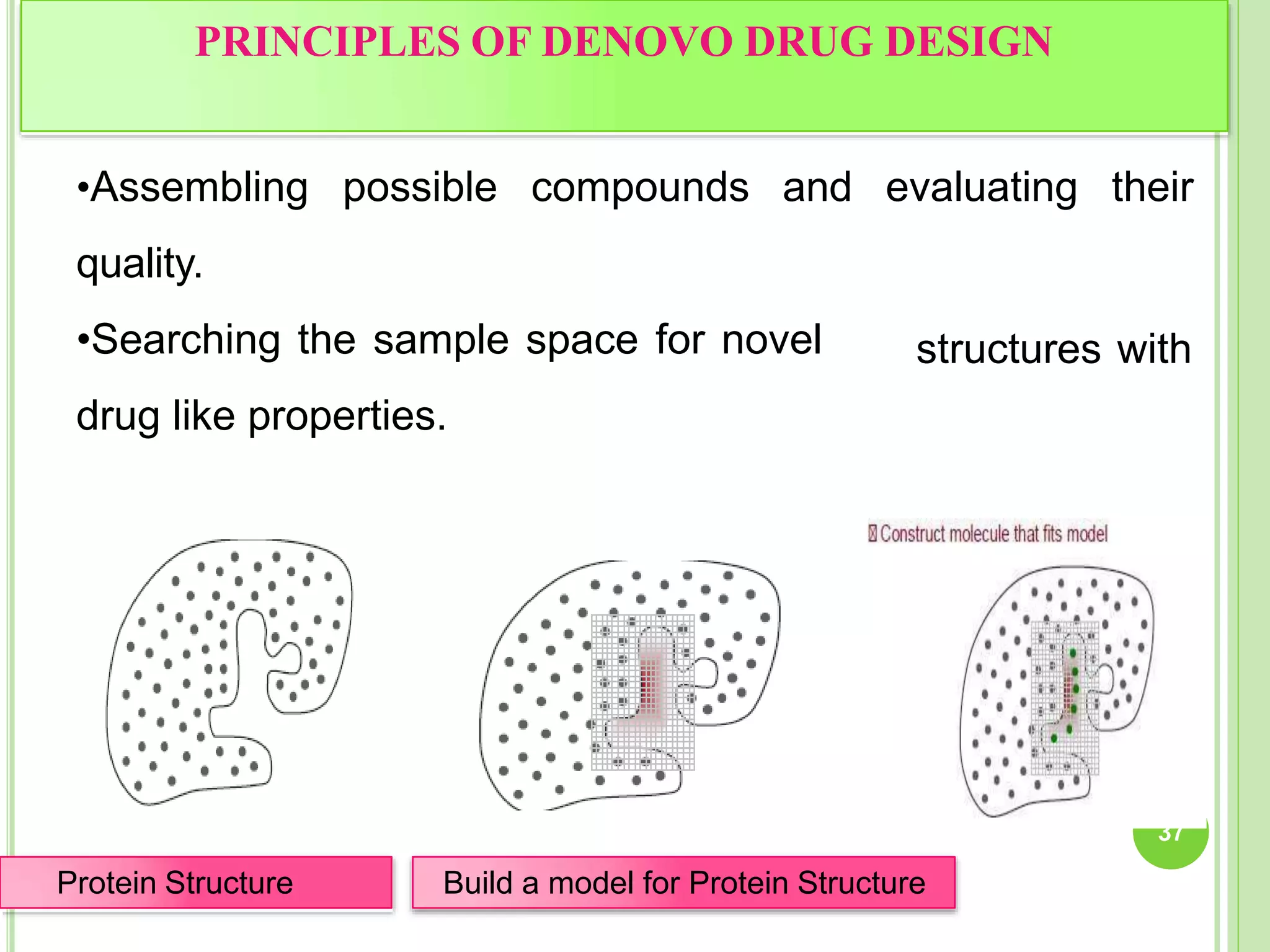
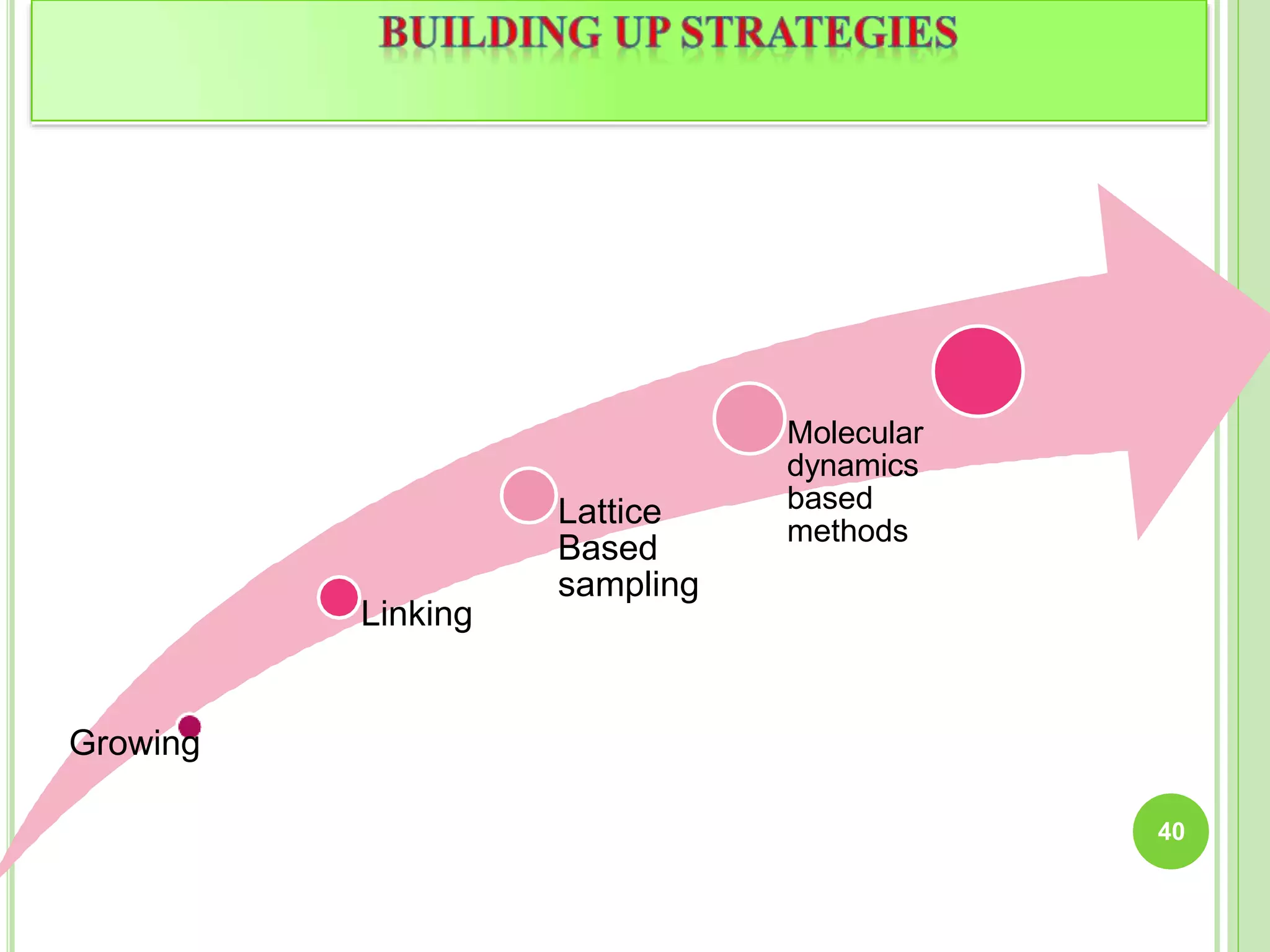
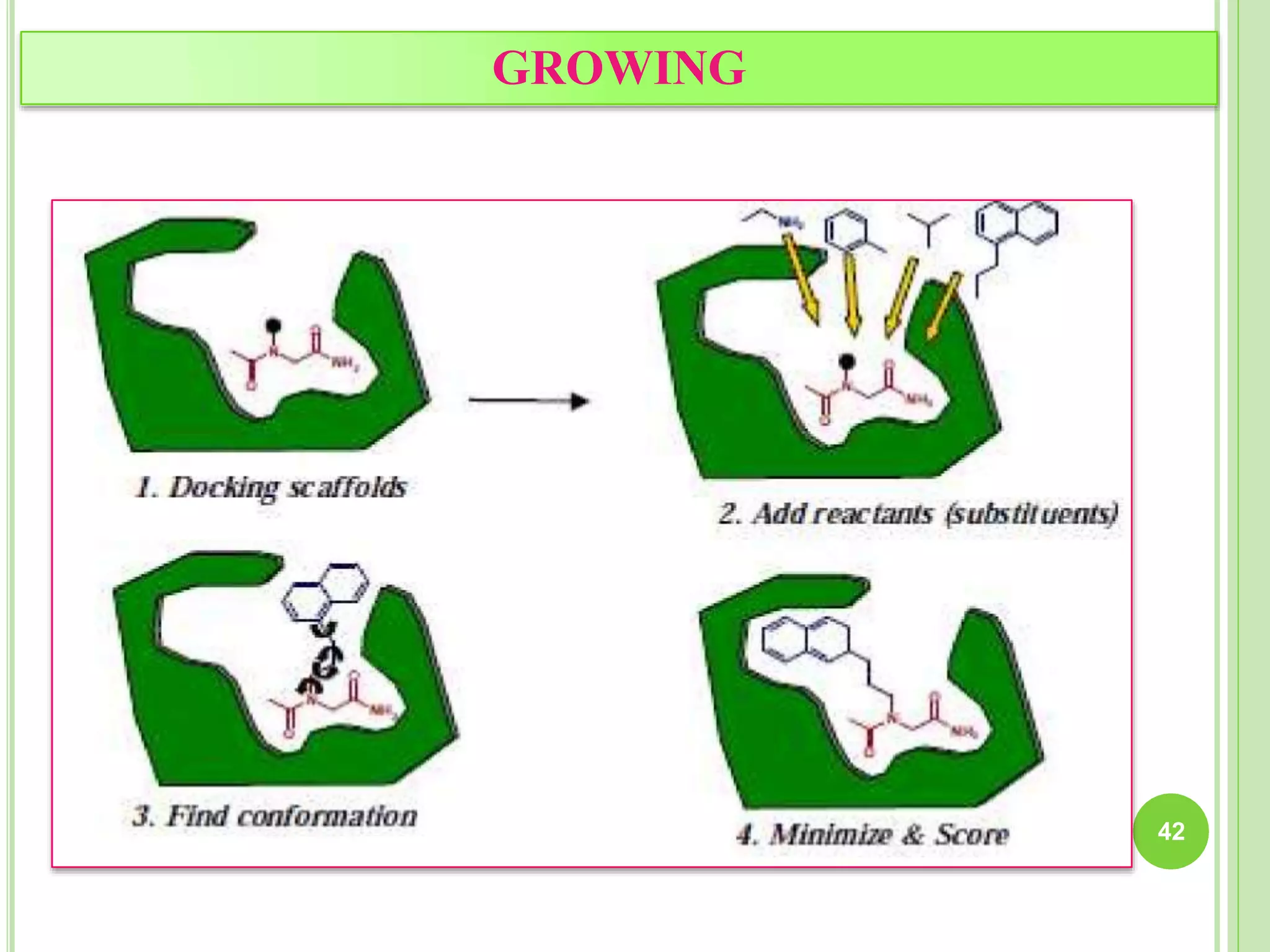
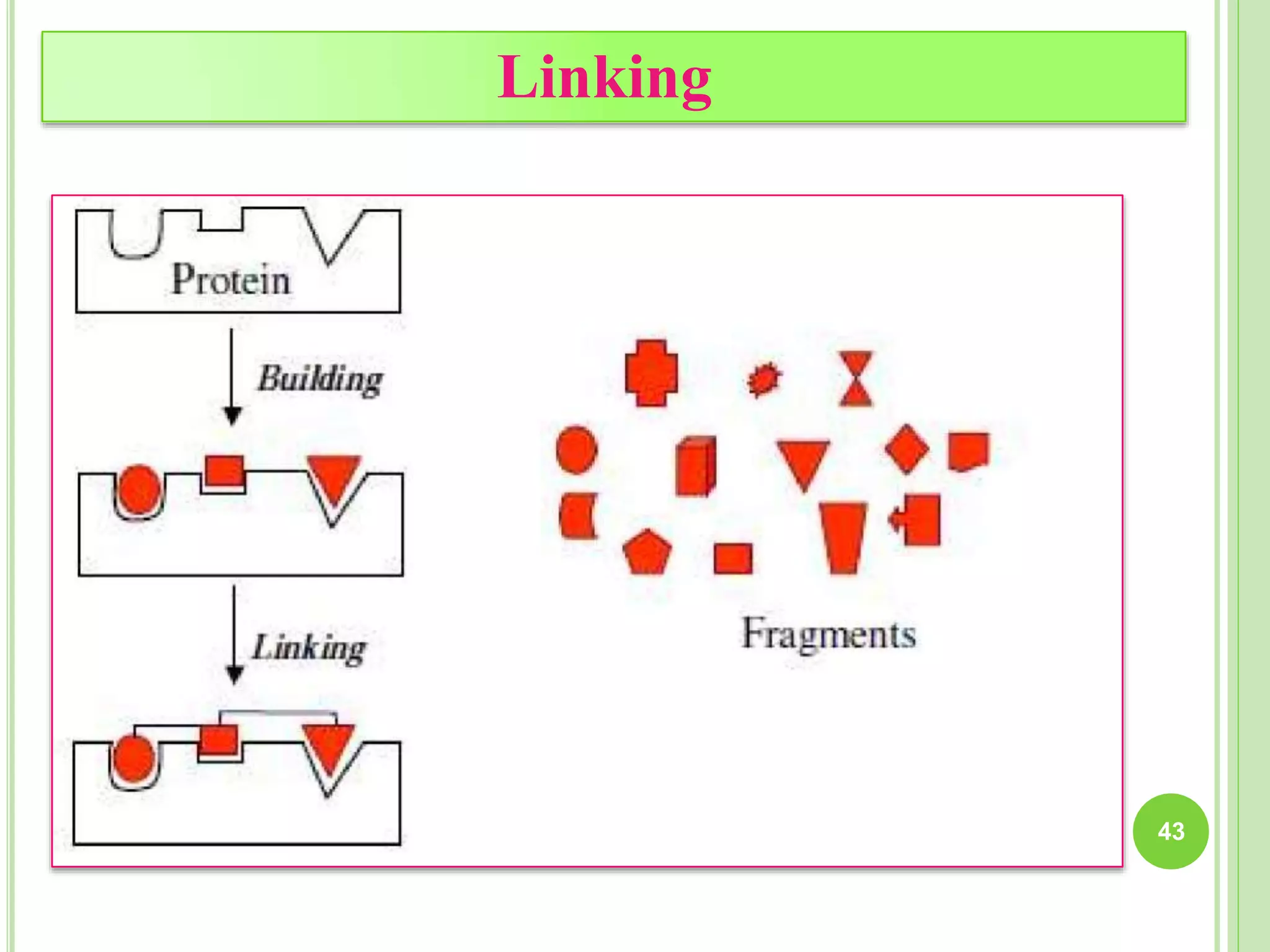
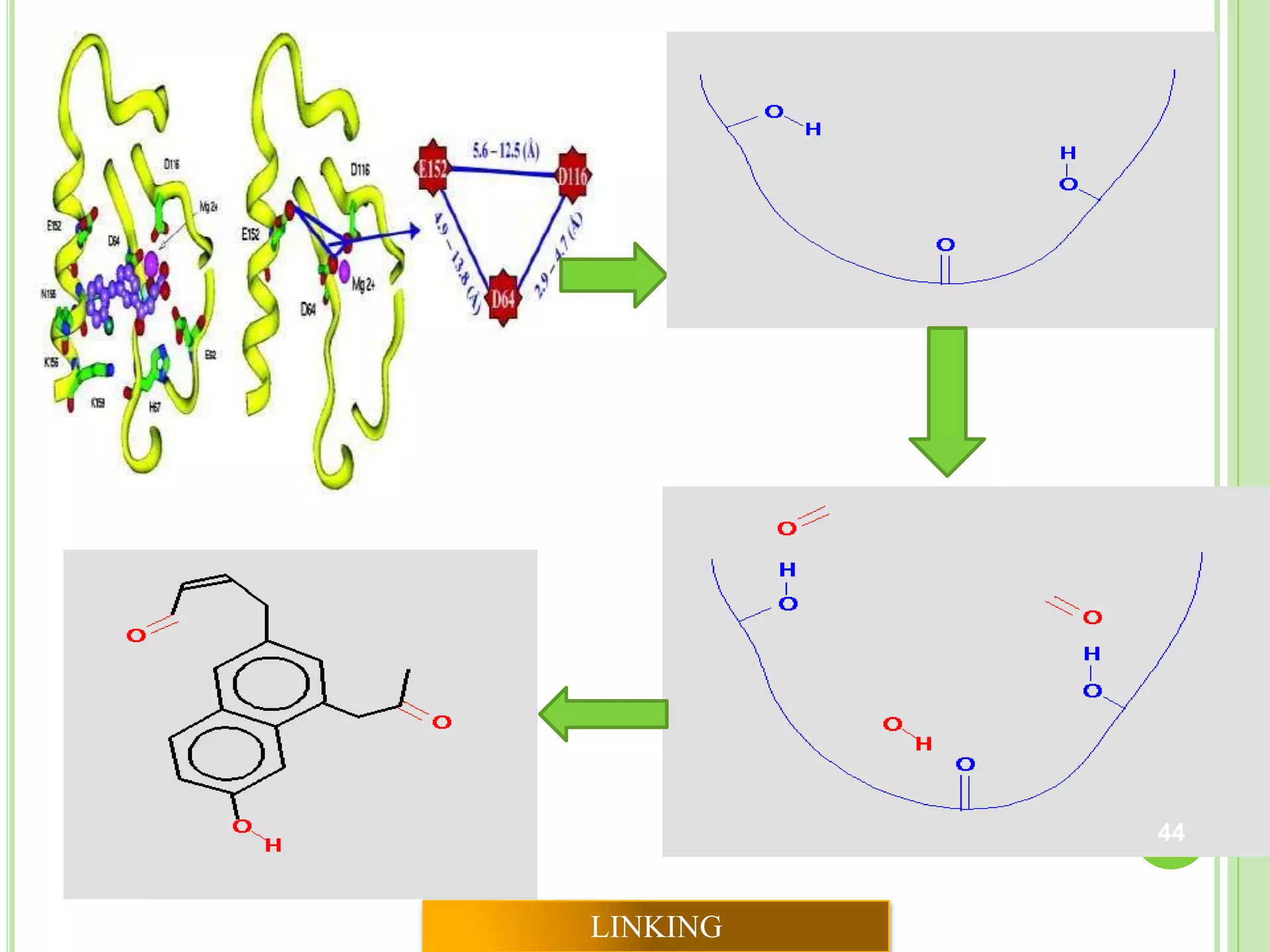
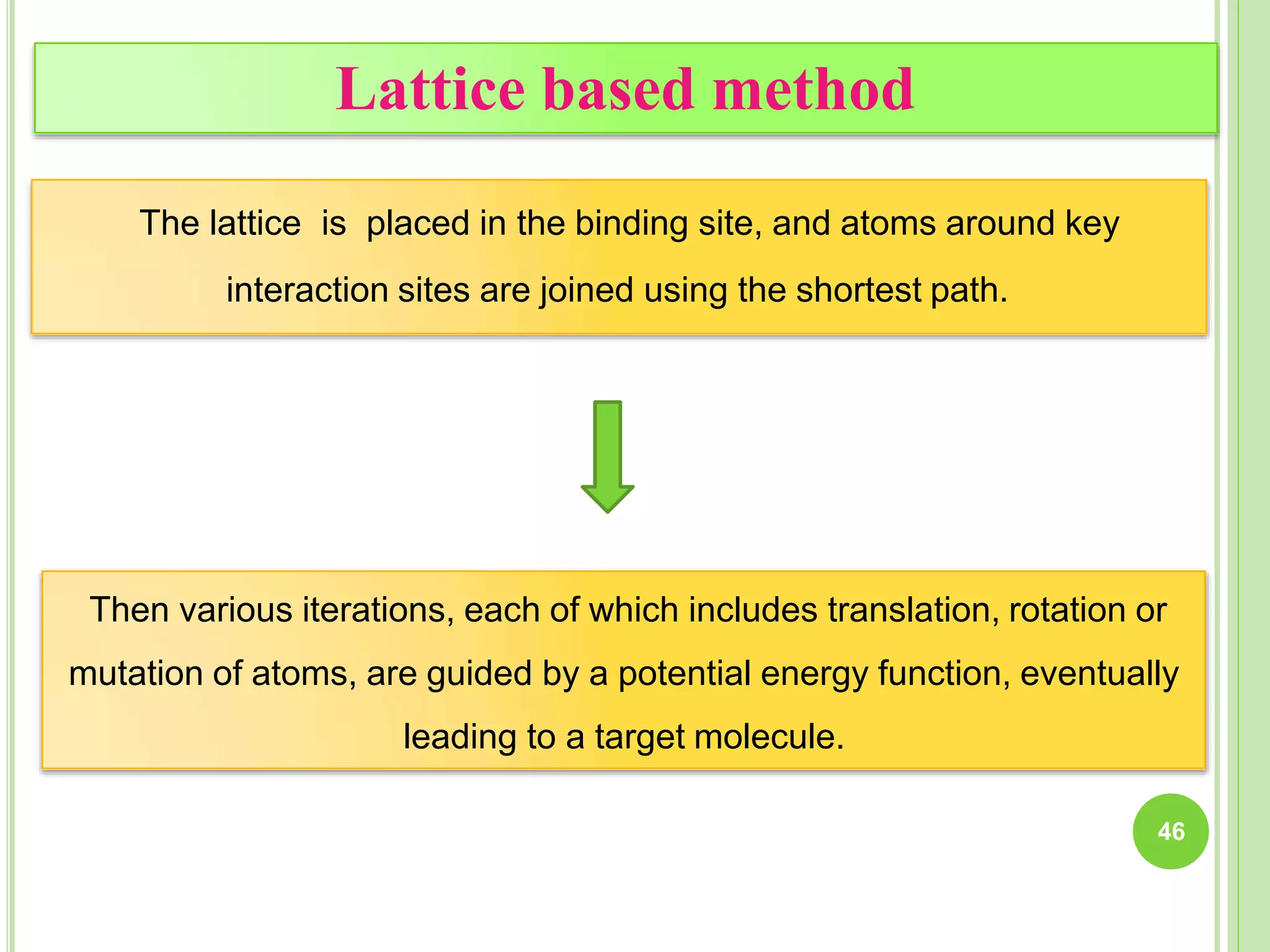
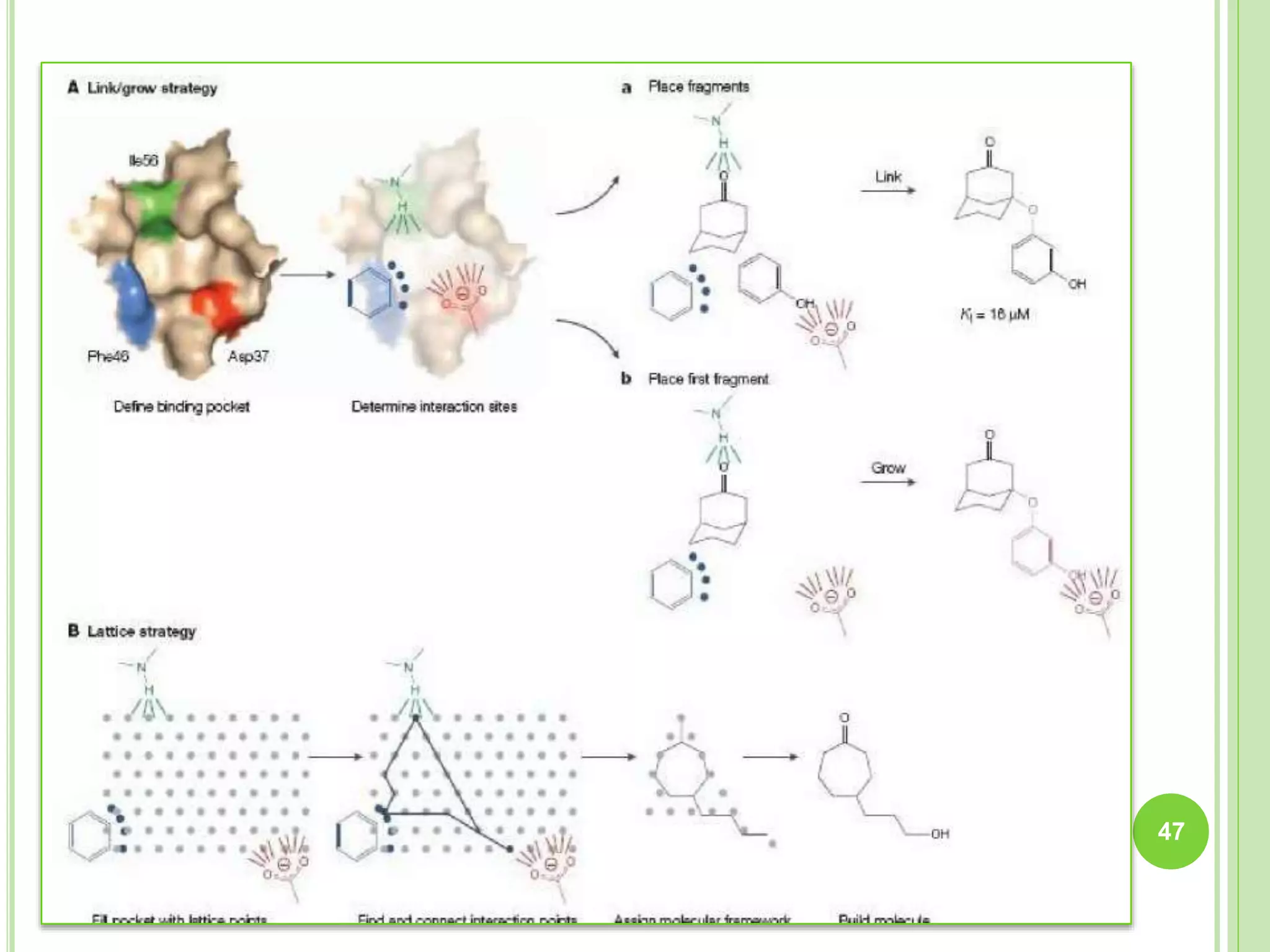
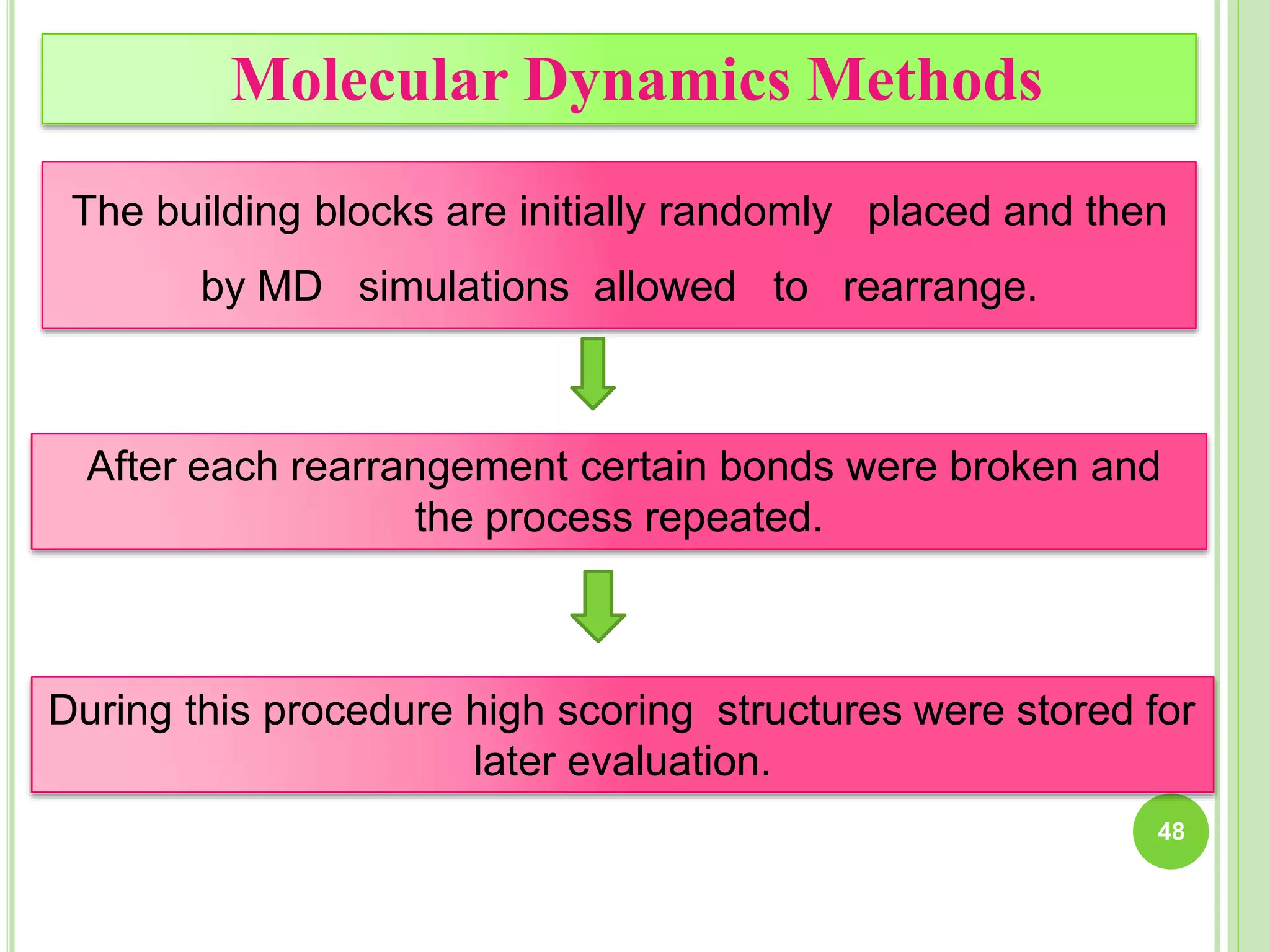
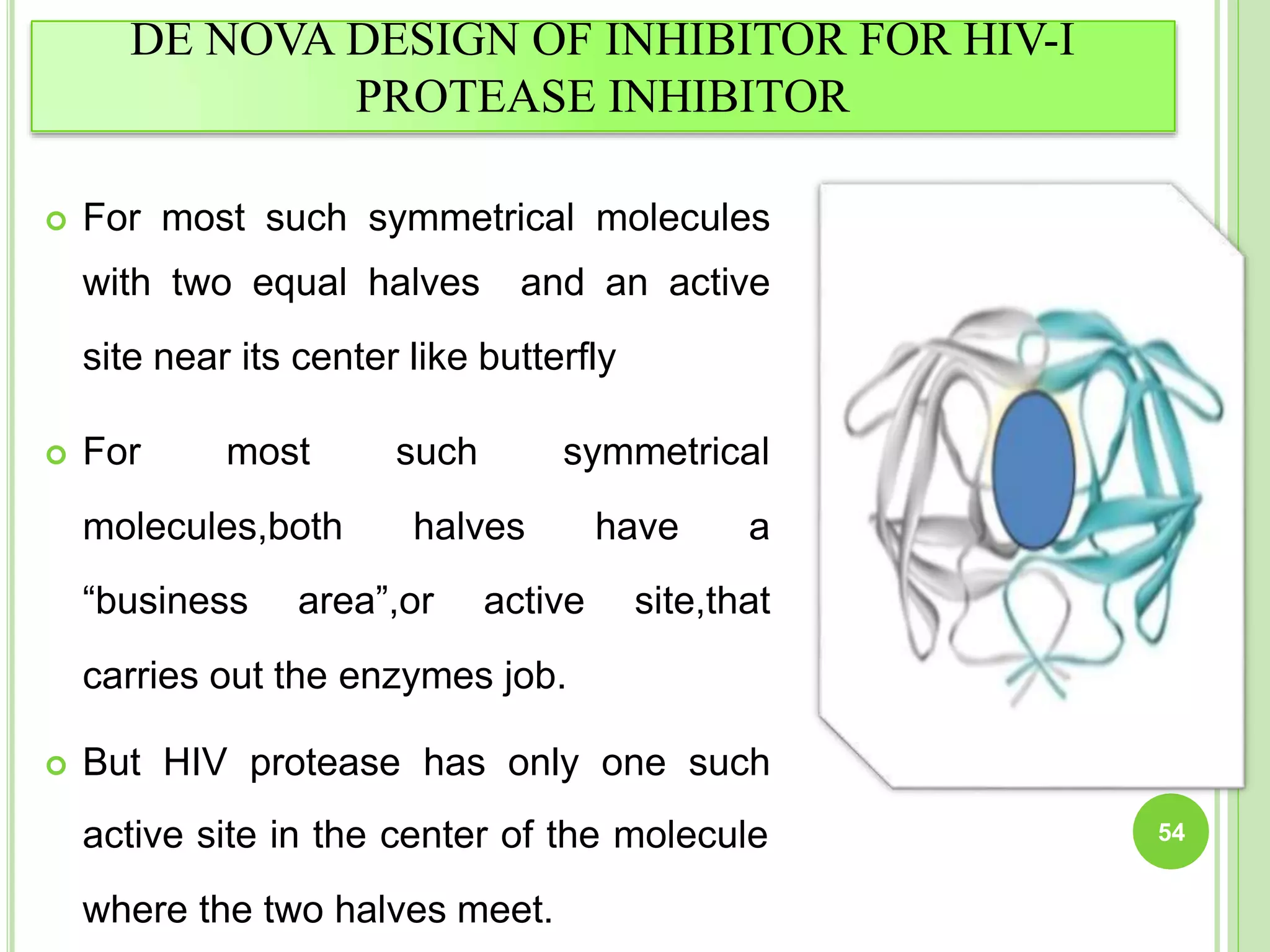
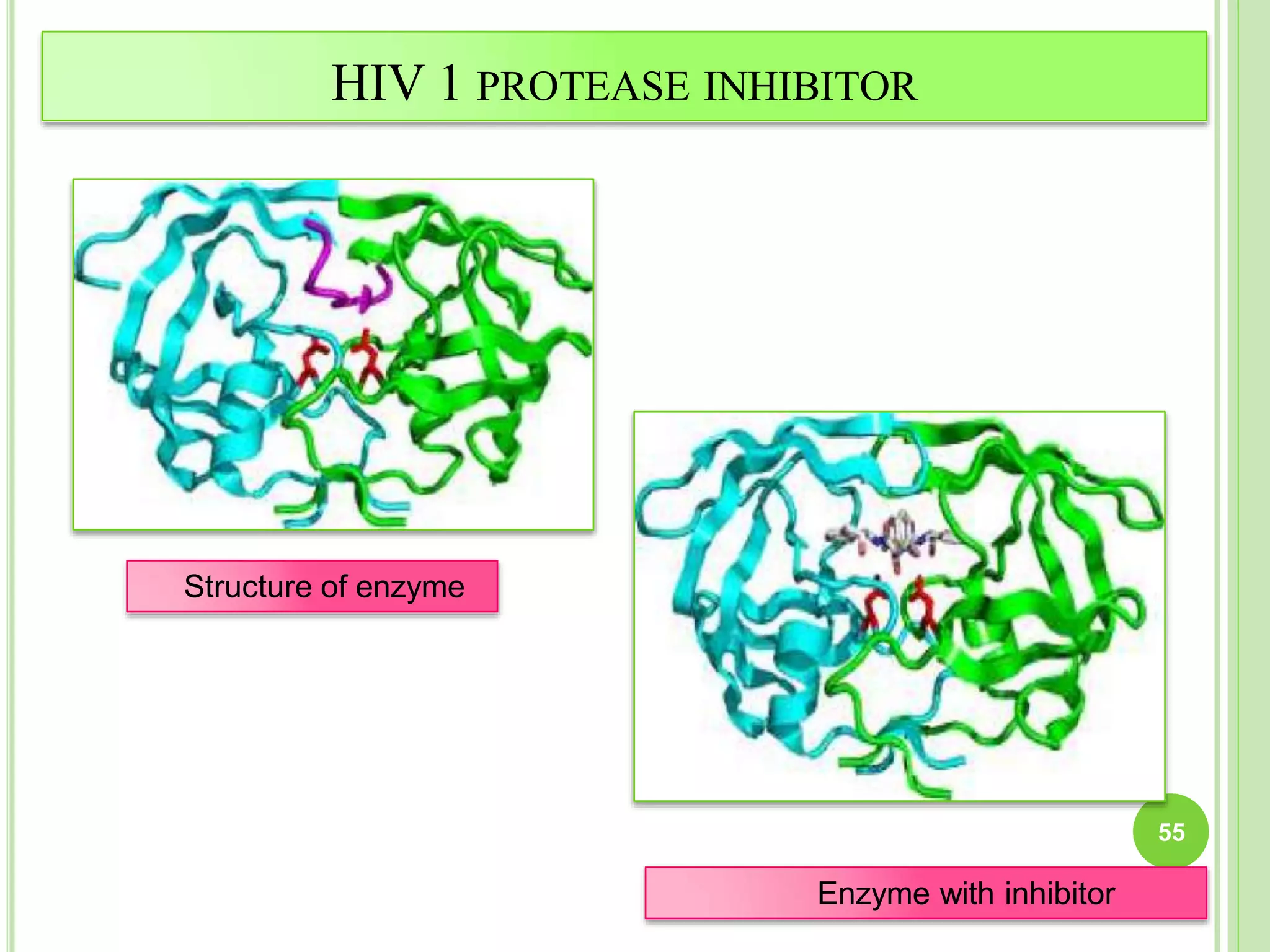
The document discusses structure-based drug design and molecular docking. It begins with introductions to drug design, drug targets, and structure-based drug design. It then describes molecular docking as a technique to predict how small molecules bind to protein targets by calculating binding affinities. The document outlines the docking process, including generating a protein's molecular surface, matching ligand and protein atoms to determine potential orientations, and scoring docked poses to identify favorable interactions. It also discusses using docking for virtual screening to identify potential drug leads from compound libraries.