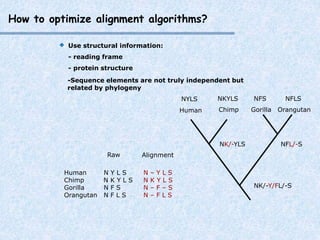
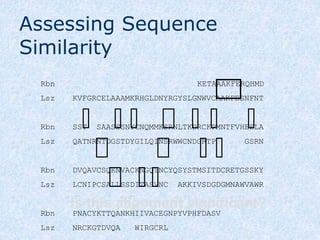
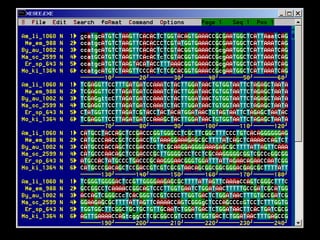
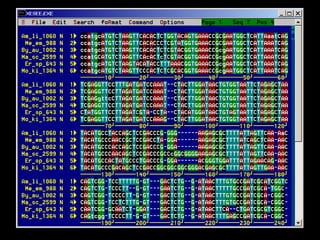
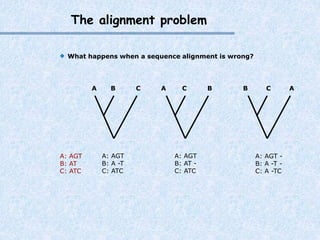
The document provides an extensive overview of sequence alignment in bioinformatics, explaining its definitions, significance, and methodologies. It details different types of alignments, including global, local, and multiple sequence alignments, as well as the algorithms used to perform them, such as Needleman-Wunsch and Smith-Waterman. It also discusses the concepts of similarity versus homology, the impact of mutations on sequence dissimilarity, and the computational complexity associated with alignment algorithms.





























![The big-O notation
•To compute a N-wise alignment, the algorithmic
complexity is something like O(c2n),
where c is a constant, and n is the number of
sequences
Example:
A pairwise alignment of two sequences [O(c2x2)], takes 1 second,
then four sequences [O(c2x4)], would take 104 seconds (2.8
hours), five sequences [O(c2x5)], 106 seconds (11.6 days), six
sequences [O(c2x6)], 108 seconds (3.2 years), seven sequences
[O(c2x7)], 1010 seconds (317 years), and so on
This is disastrous!](https://image.slidesharecdn.com/introductiontosequencealignment-151201093826-lva1-app6892/85/Introduction-to-sequence-alignment-33-320.jpg)