Downloaded 108 times

![ICH PARTNERS
EFPIA[European Federation of pharmaceutical
Industries and Associations]
JPMA[Japan Pharmaceutical Manufactures
Association]
PhRMA[Pharmaceutical Research and Manufactures
Of America]
MHLW[Ministry Of Health Labor Welfare]Japan
USFDA
EU regulators](https://image.slidesharecdn.com/ichguidelinesandprotocols-140929003224-phpapp02/75/Ich-guidelines-and-protocols-2-2048.jpg)











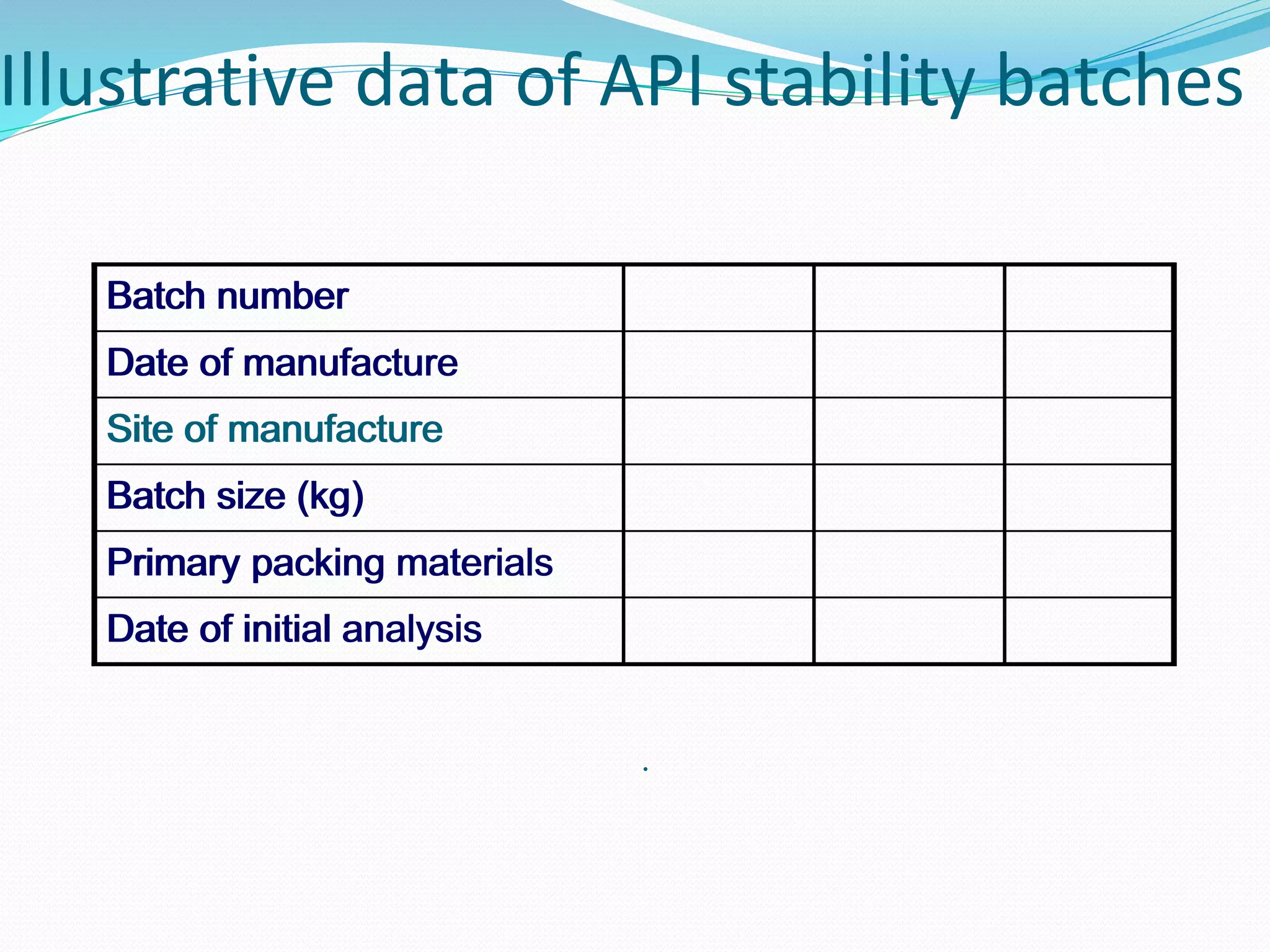
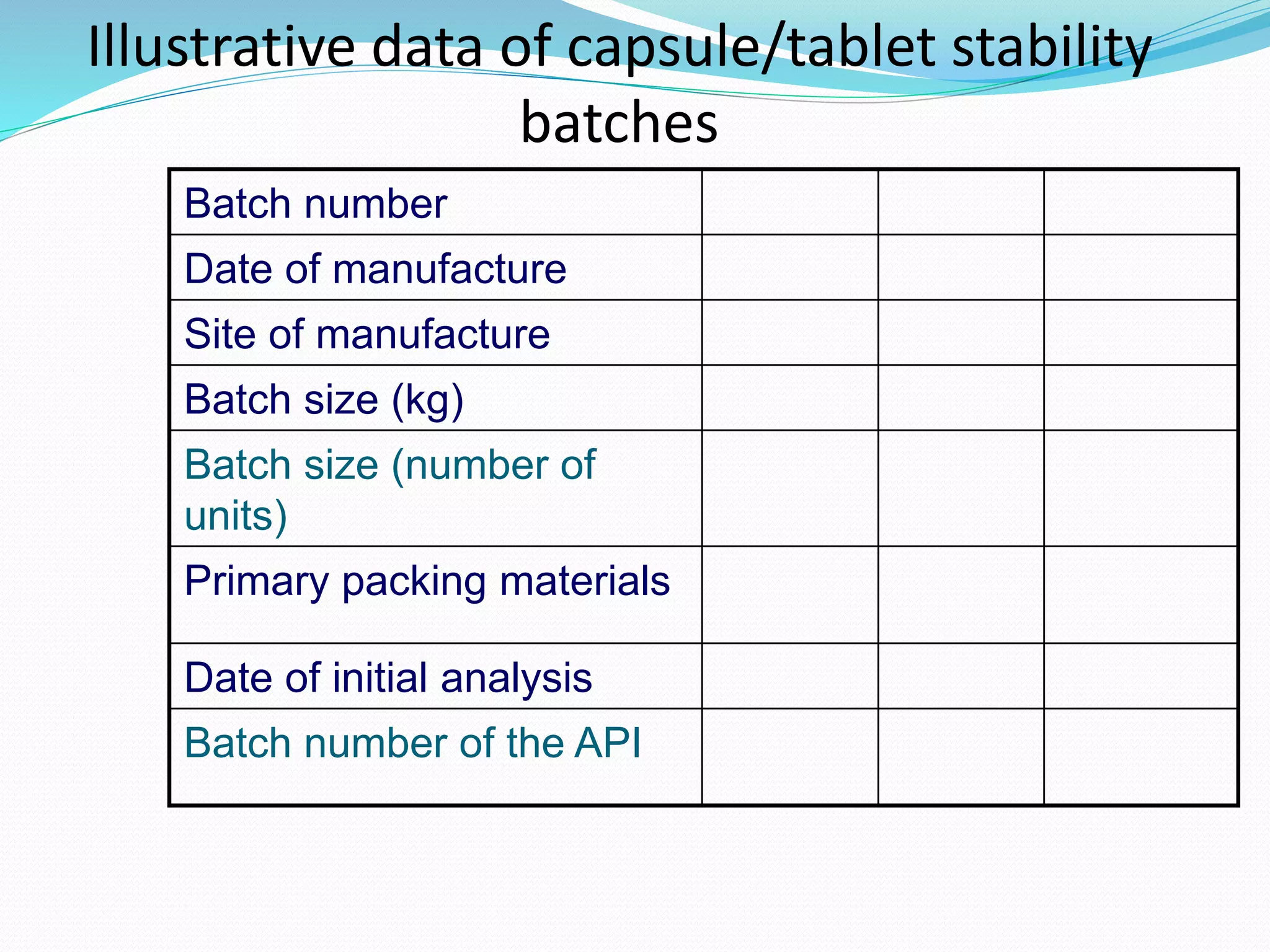







This document discusses ICH guidelines for stability testing and protocols. It provides an overview of the ICH partners that develop guidelines and describes some of the key ICH guidelines related to quality, safety, and efficacy. It then focuses on ICH guideline Q1 which provides recommendations for stability testing of new active pharmaceutical ingredients and finished pharmaceutical products, including stress testing, selection of batches, storage conditions, and photo stability testing. The document also discusses bracketing and matrixing designs for stability testing and outlines what should be included in a stability protocol and report.











































































































