This document discusses regulatory aspects of stability testing in Europe. It provides an overview of the objectives and requirements for stability testing of pharmaceutical ingredients and products containing them. The key points are:
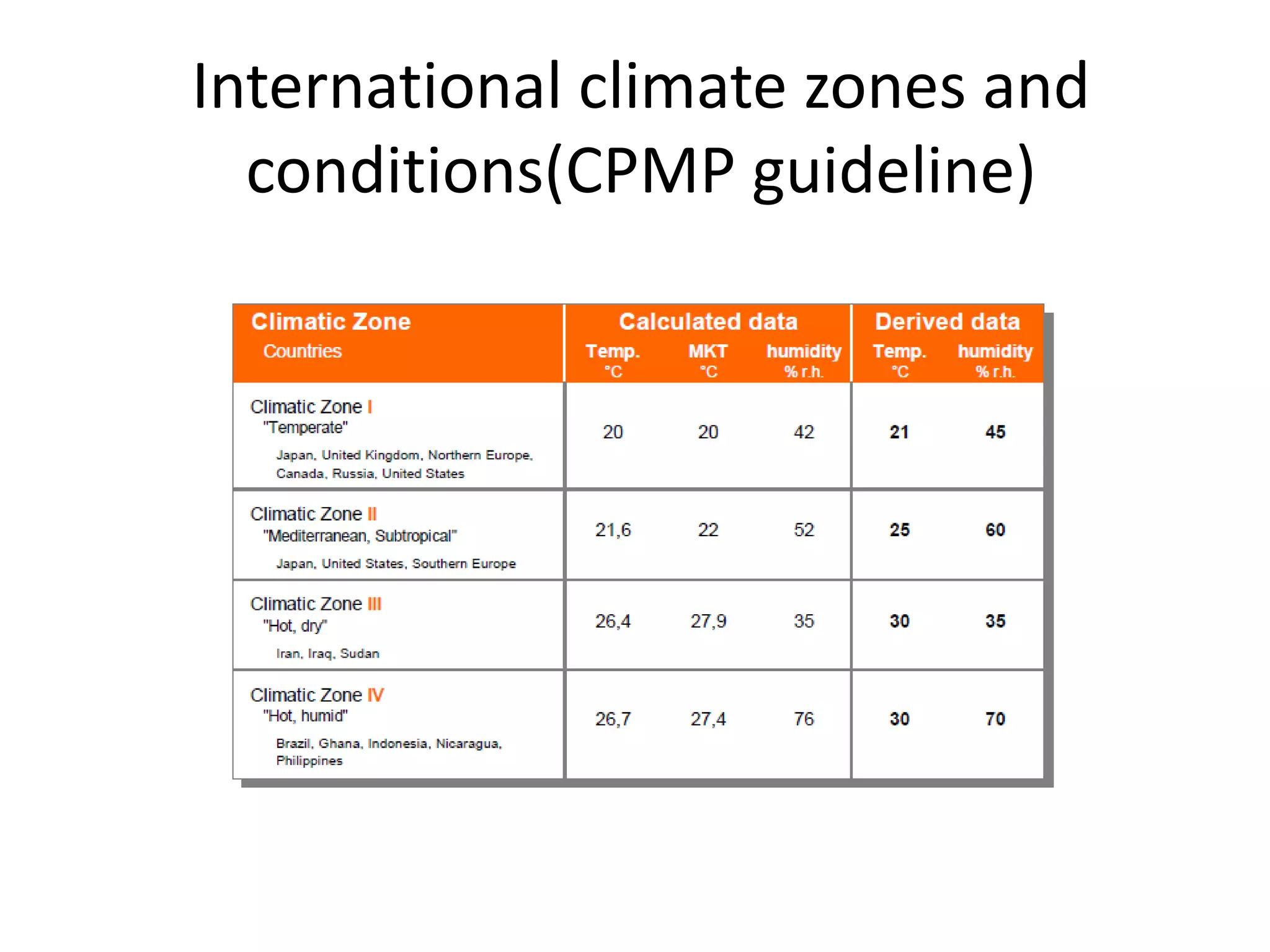
1) Stability testing aims to establish appropriate storage conditions and shelf lives by evaluating how quality varies over time under different environmental factors like temperature, humidity, and light.
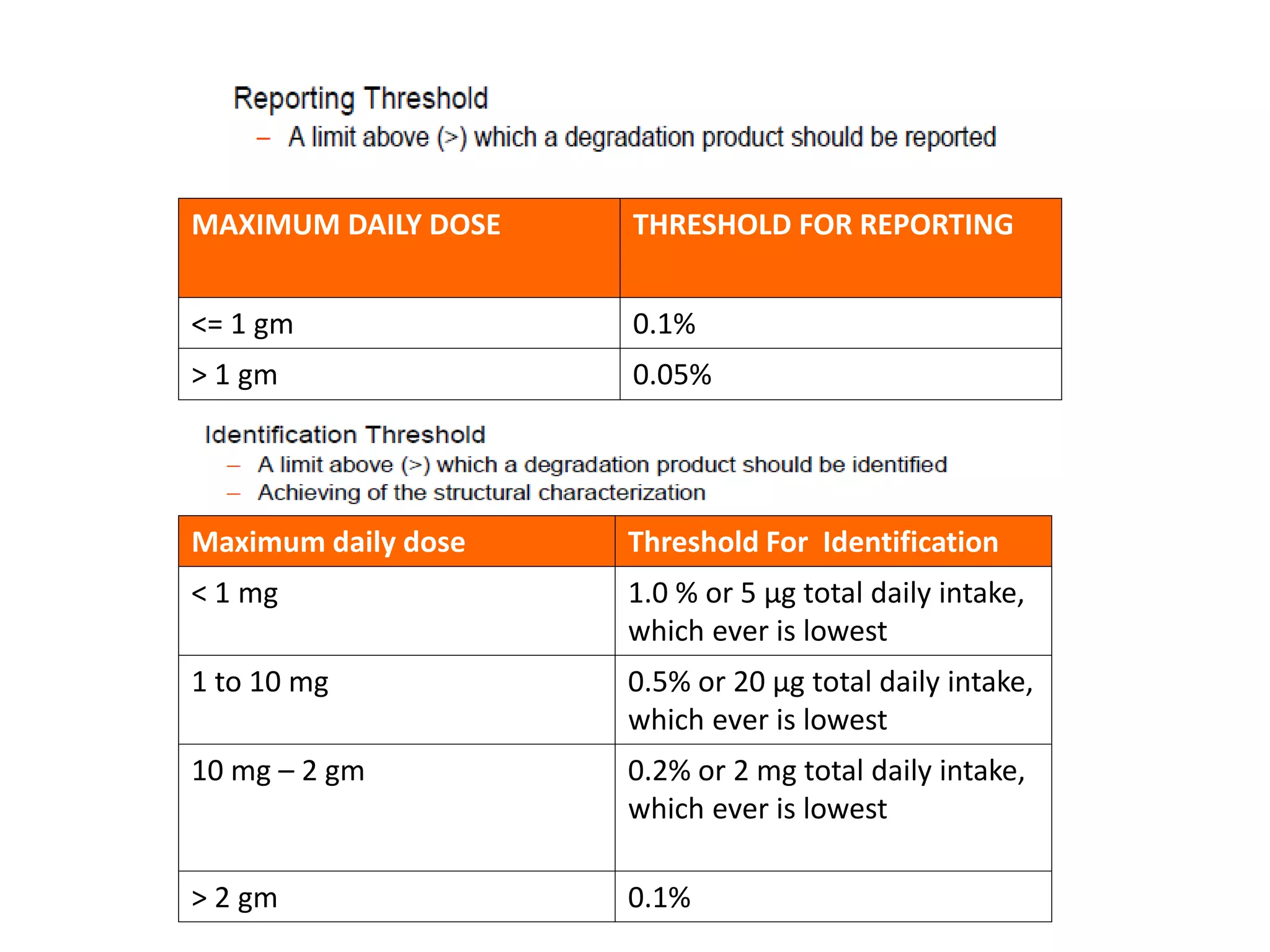
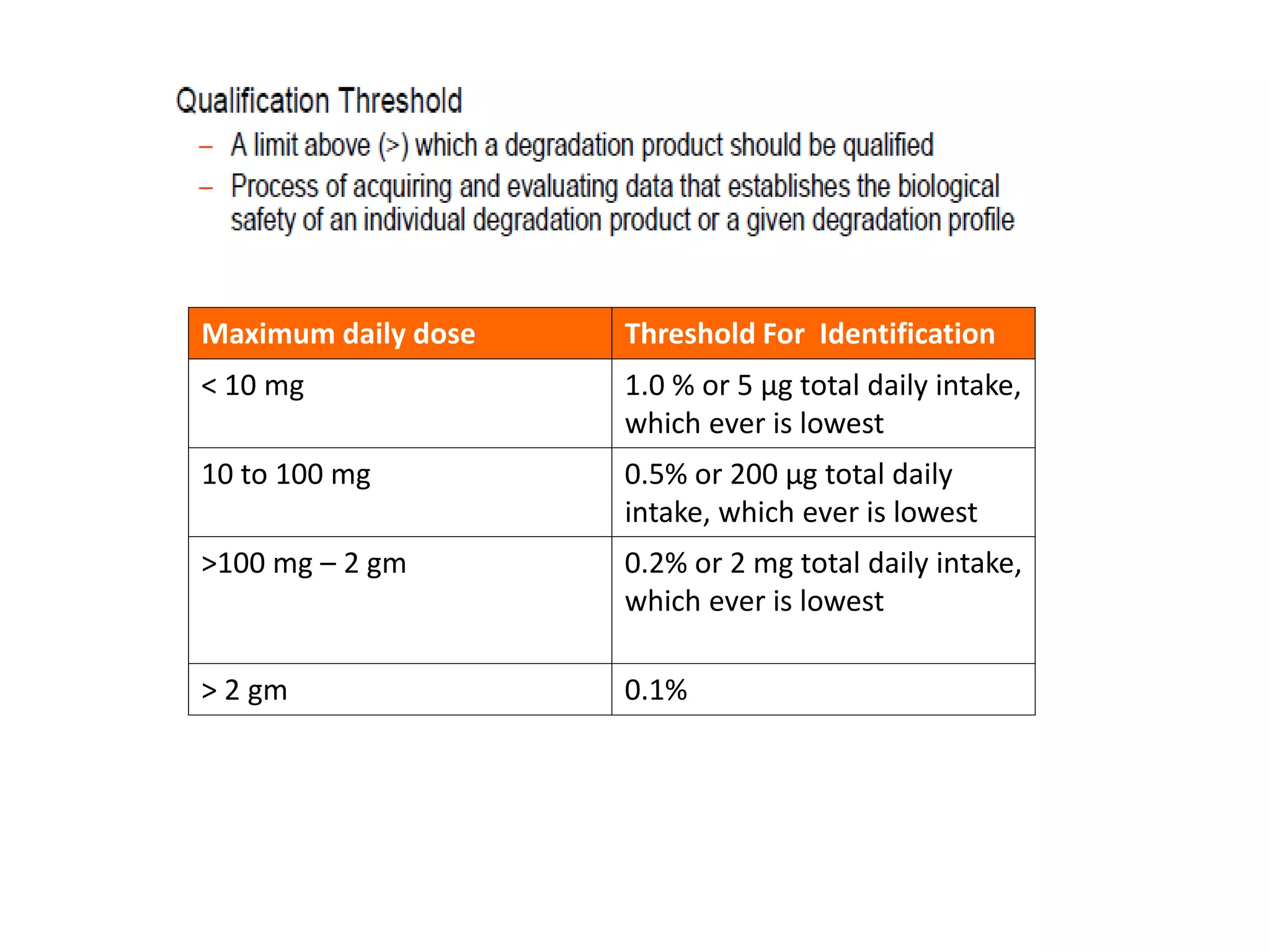
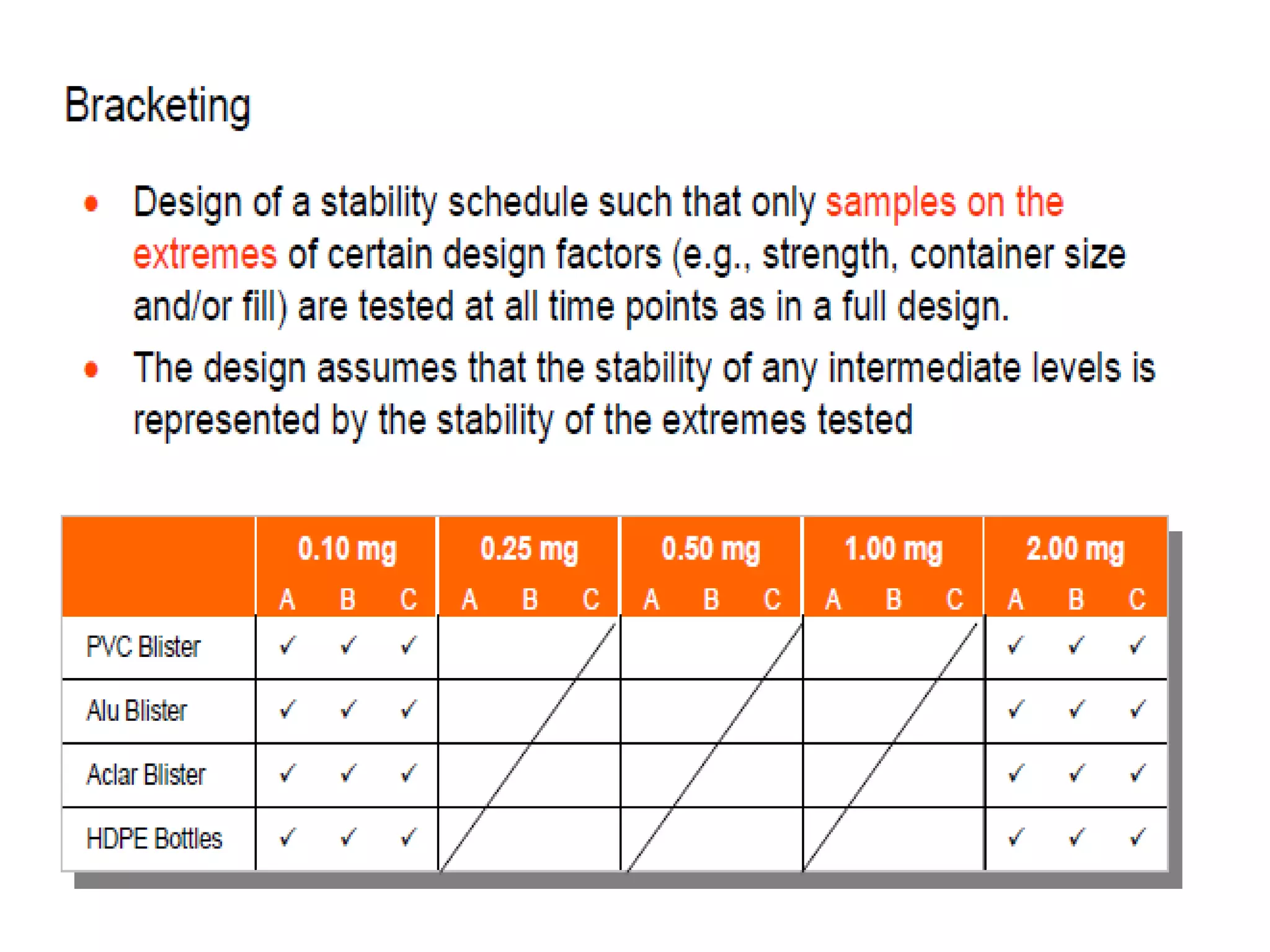
2) Testing is required for both active ingredients and finished products to justify storage conditions, retest periods, and shelf lives. It involves evaluating physical, chemical, microbial, and other changes over time under various conditions.
3) Guidelines from the European CPMP and ICH specify design, conduct, evaluation, and reporting of stability studies to support regulatory submissions in