Downloaded 743 times












![Light source used for photostability
testing:
Option 2:the cool white fluorescent and near
ultraviolet lamp.
1. A cool white fluorescent lamp designed to produce
an output similar to that specified in ISO.
[D65 10977(1993)]
2. A near UV fluorescent lamp having a spectral
distribution from 320 nm to 400 nm](https://image.slidesharecdn.com/ichguidelinesforstabilitytesting-190928063906/85/ICH-Guidelines-17-320.jpg)
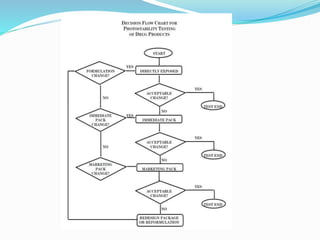

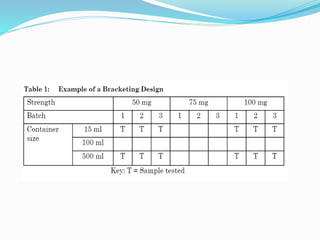



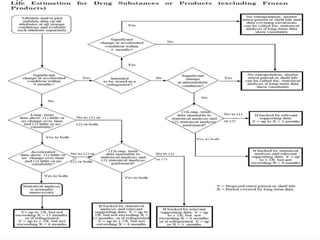





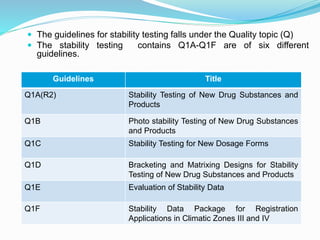
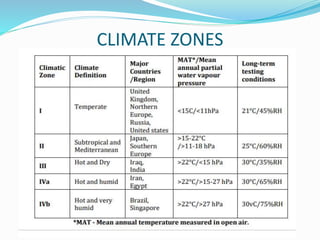
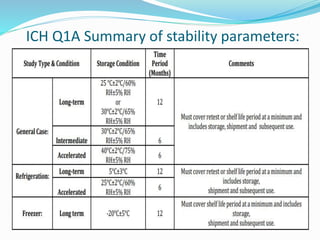
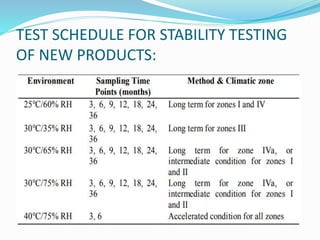
The document discusses the objectives and guidelines of the International Council for Harmonization (ICH) for stability testing of pharmaceutical products. It provides an overview of the key ICH guidelines for stability testing (Q1A-Q1F) and describes the principles of stability testing including establishing re-test periods and shelf lives. It also discusses the different types of stability testing, protocols, study designs like bracketing and matrixing, and key parameters for evaluation.



































































