Downloaded 1,068 times







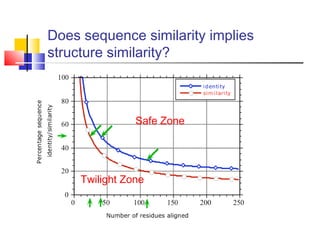
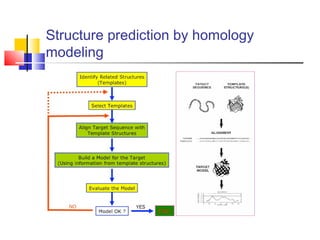
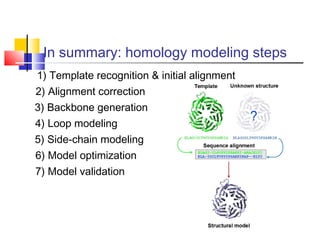


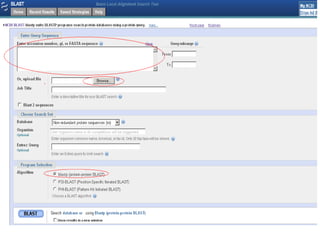
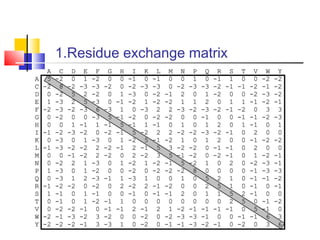
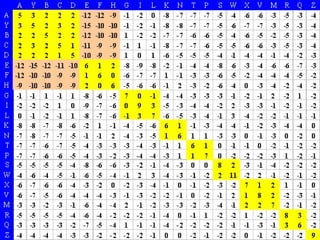


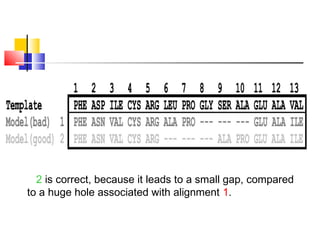
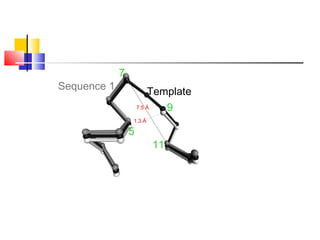


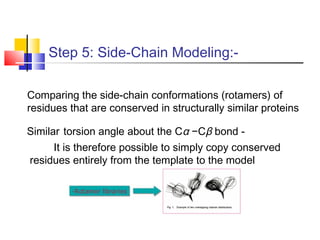
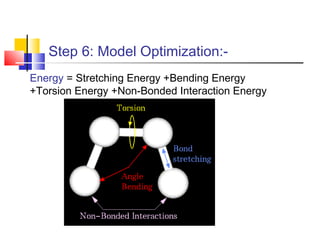

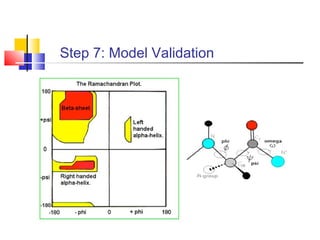



Homology modeling is a technique used to predict the 3D structure of a protein based on the alignment of its amino acid sequence to known protein structures. It relies on the observation that structure is more conserved than sequence during evolution. The key steps in homology modeling include: 1) identifying a template structure through sequence alignment tools like BLAST, 2) correcting any errors in the initial alignment, 3) generating the protein backbone based on the template structure, 4) modeling any loops or missing regions, 5) adding side chains, 6) optimizing the model structure energetically, and 7) validating that the final model matches the template structure and has correct stereochemistry. Homology modeling is useful for applications like structure-based drug design











































































![Bio info statistical-methods[1]](https://cdn.slidesharecdn.com/ss_thumbnails/bioinfo-statisticalmethods1-150123025112-conversion-gate02-thumbnail.jpg?width=640&height=640&fit=bounds)



















