Downloaded 204 times




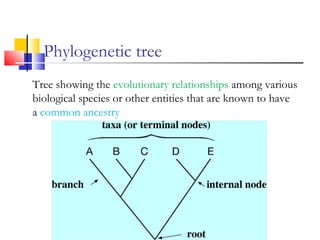
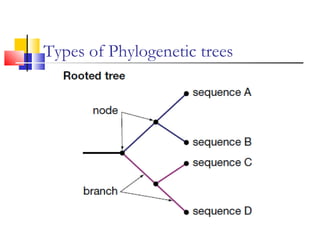
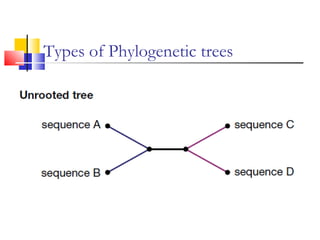
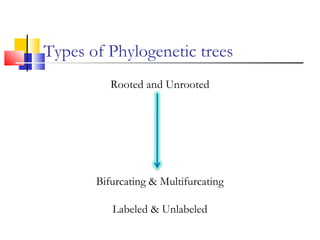


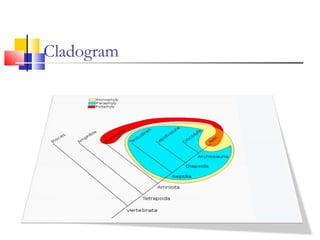
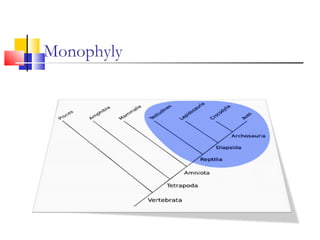
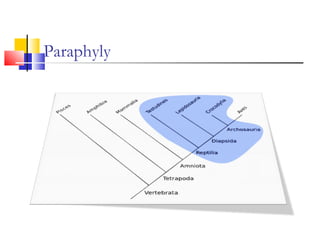



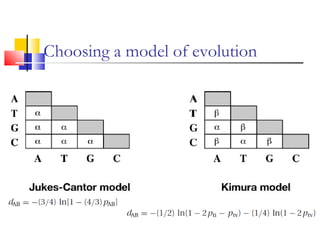






This document discusses phylogenetic studies and the construction of phylogenetic trees. It notes that fossil records are unreliable, so phylogenetic trees are primarily based on molecular sequencing data and morphological data. There are several assumptions made in phylogenetic analysis, including that sequences are homologous, phylogenetic divergence is bifurcating, and each position in a sequence evolved independently. The document outlines different types of phylogenetic trees, steps in phylogenetic analysis like choosing molecular markers and tree building methods, and criteria for assessing the reliability of phylogenetic trees.































































































