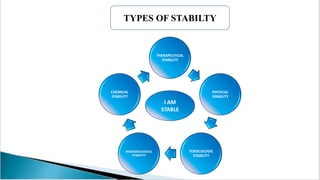
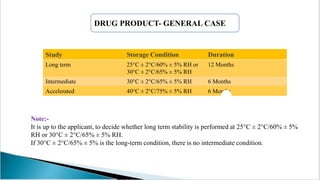
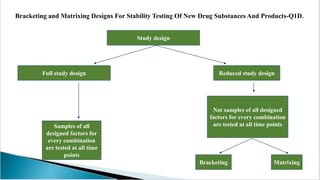
The document summarizes ICH guidelines for stability studies of new drug substances and products. It discusses the objectives and scope of stability testing, including providing evidence of a drug's quality over time under various environmental conditions to establish storage requirements and shelf life. The types of stability testing include chemical, physical, microbiological, therapeutic, and toxicological. Testing is conducted over various time periods and storage conditions as outlined in the ICH Q1A-Q1F guidelines. Evaluation of stability data includes assessing parameter results and using statistical analyses to determine a product's retest period or shelf life.