
Downloaded 13 times



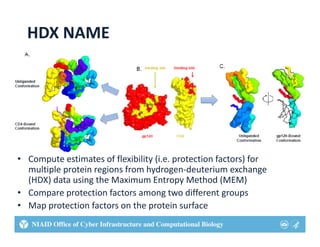








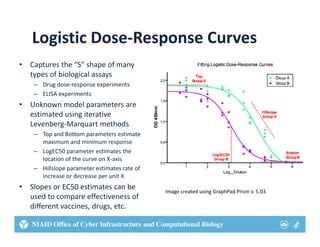


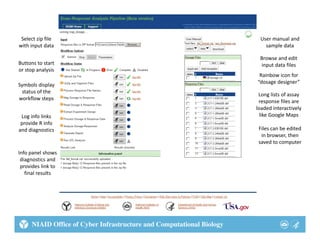

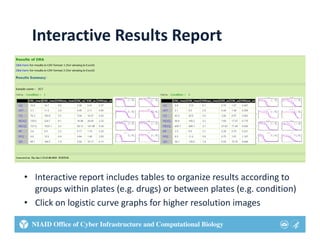
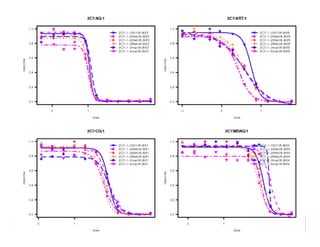


This document describes two web tools that were created using R to automate biostatistics workflows: HDX NAME and DRAP. HDX NAME analyzes hydrogen-deuterium exchange mass spectrometry data to estimate protein flexibility. It computes protection factors, compares groups, and maps results to protein structures. DRAP fits logistic dose-response curves to drug screening data from multiple plates. It automates curve fitting, compares results, and exports summaries. Both tools were created with R on the backend for analysis and web interfaces for usability. This allows researchers to perform complex analyses without programming expertise.























































































