Downloaded 141 times

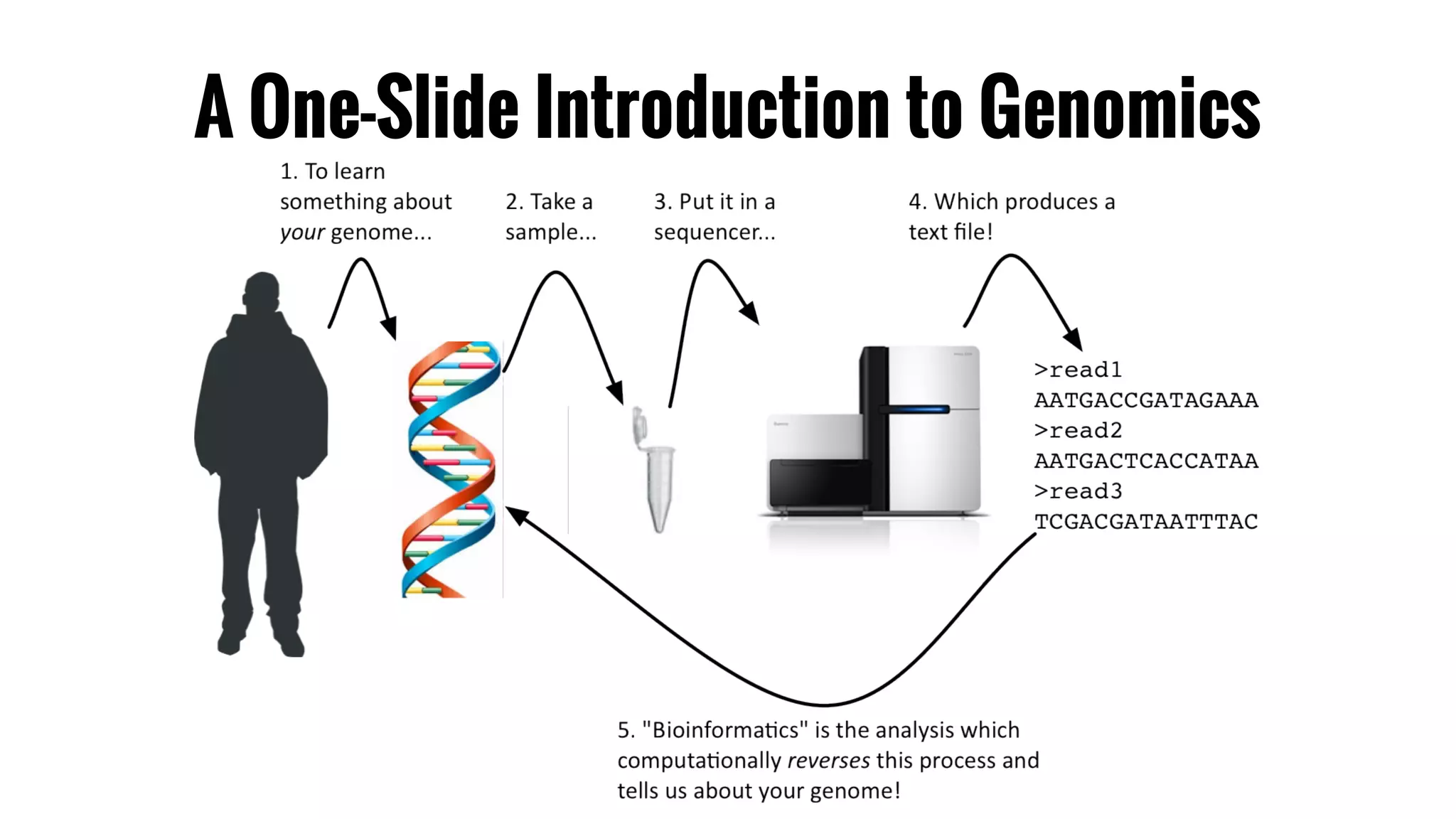

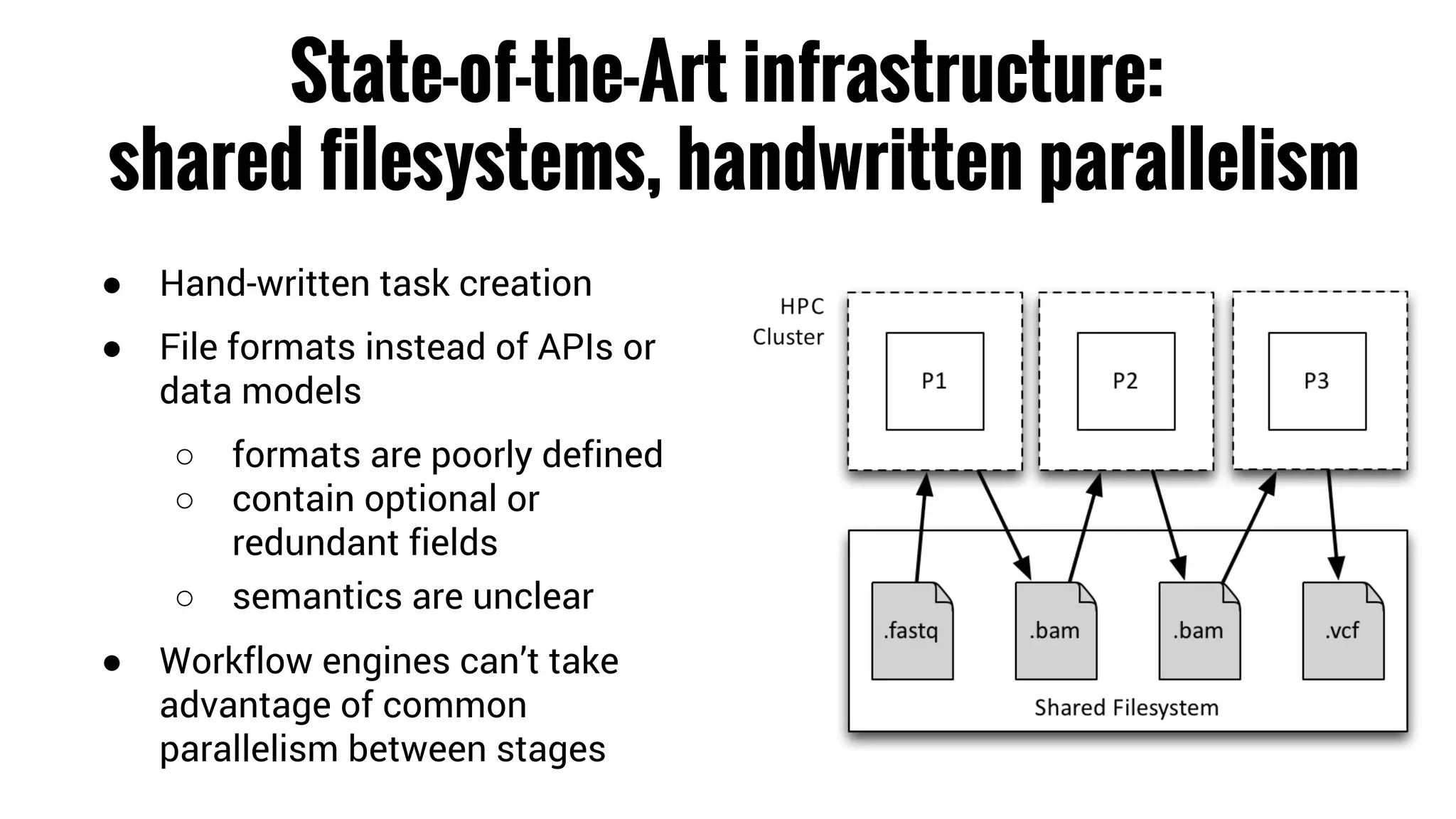


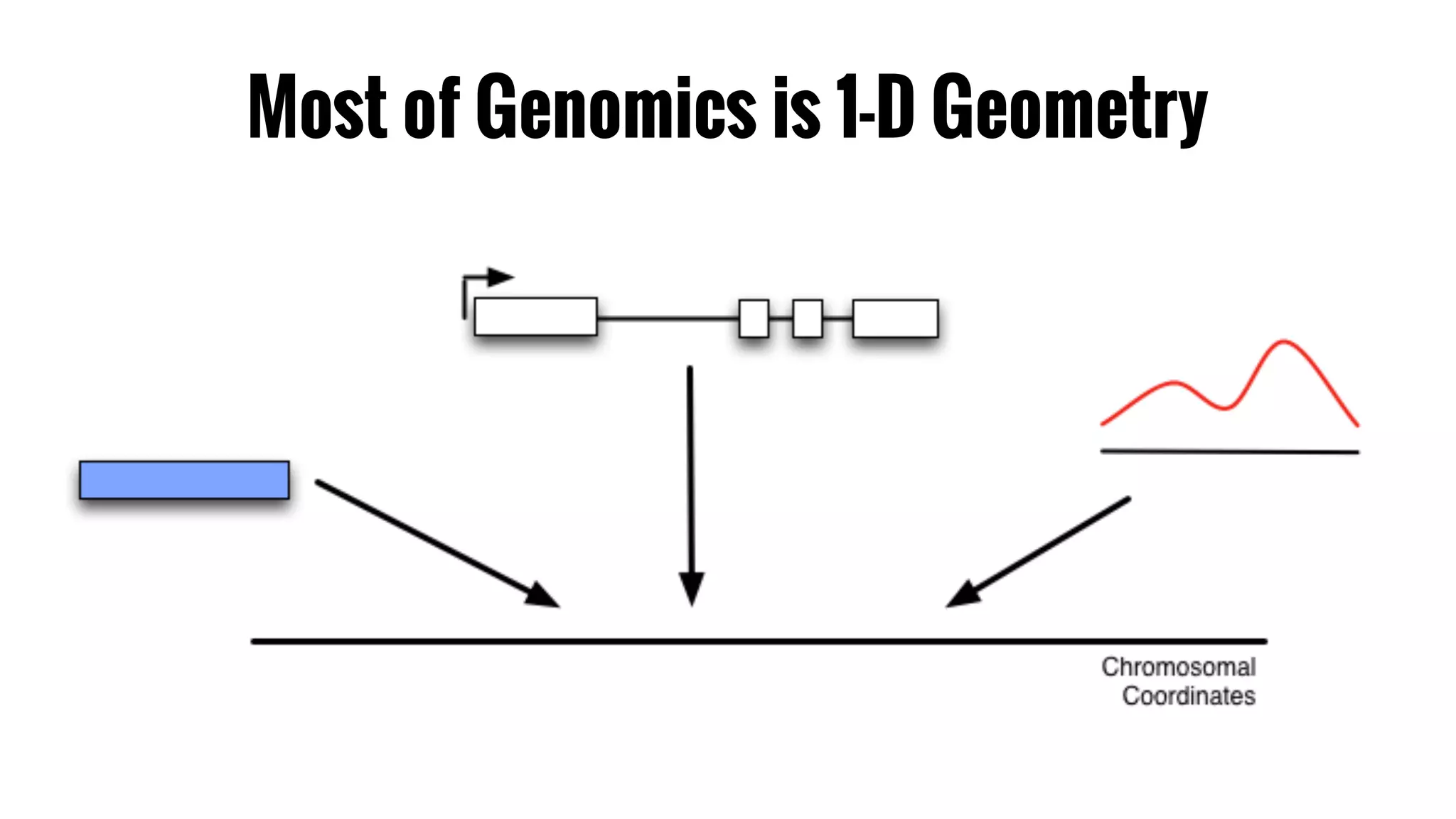

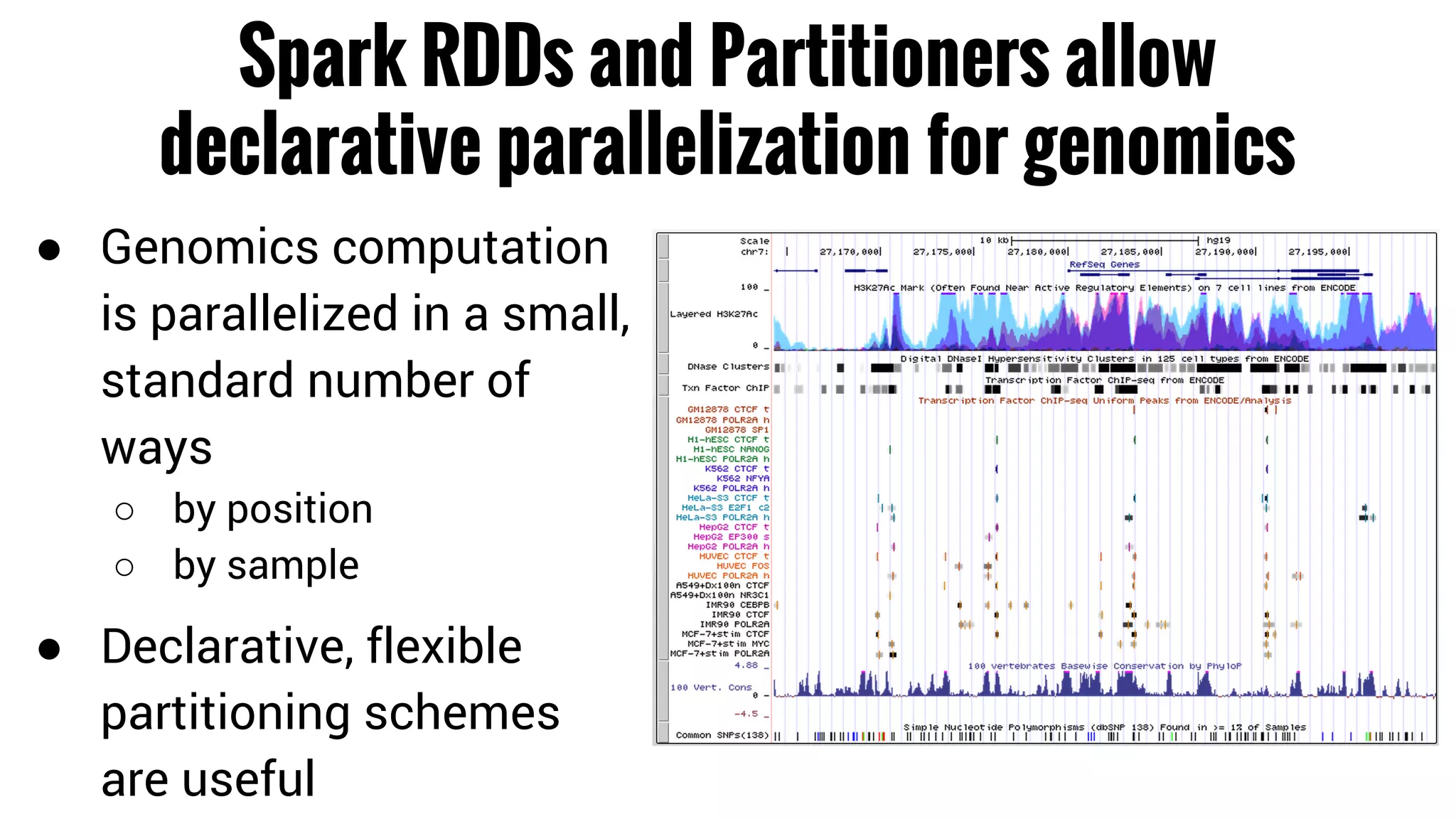
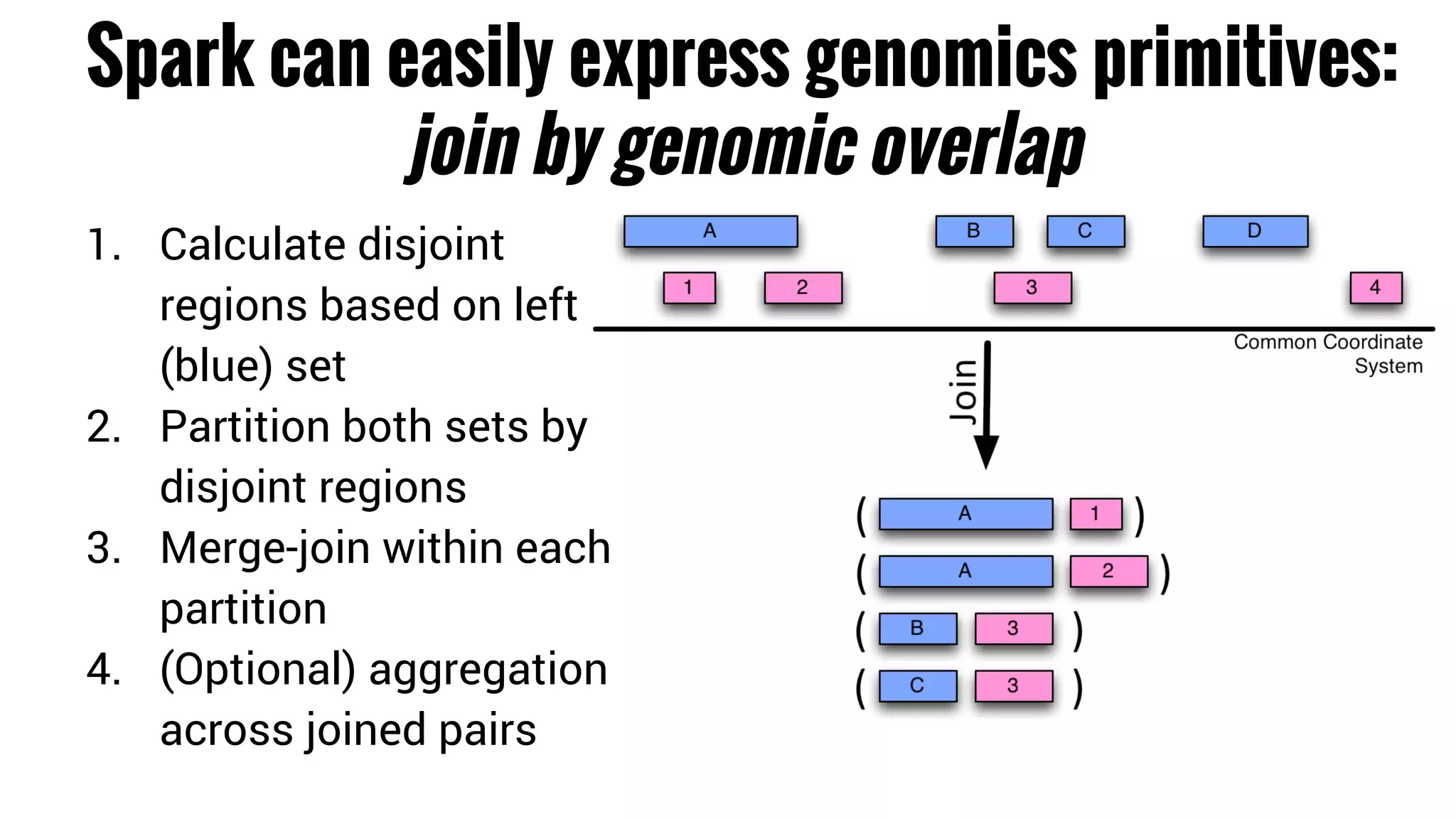


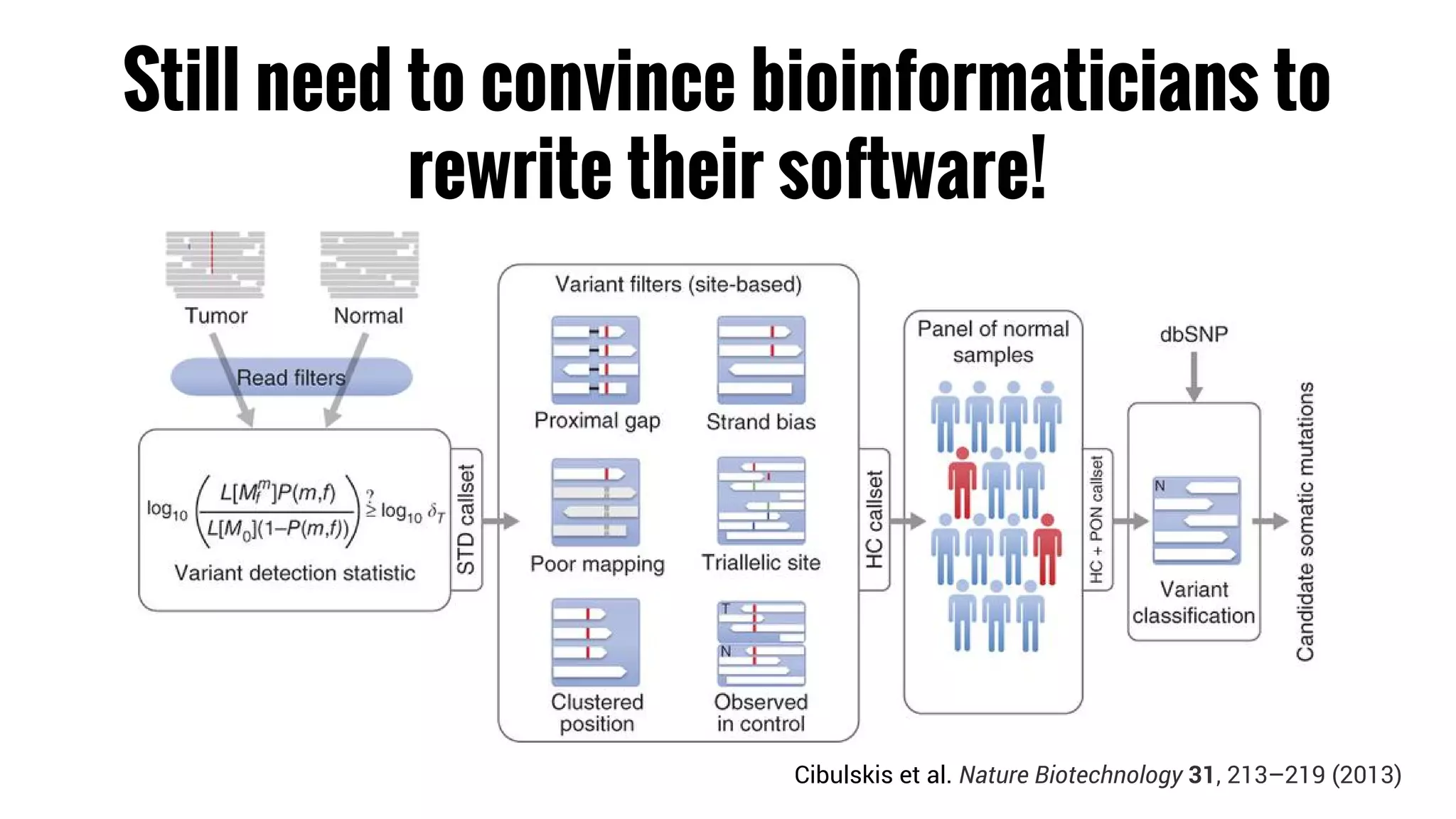




The document discusses the potential of using Apache Spark for bioinformatics, specifically genomics, highlighting its advantages in handling batch processing and workflows. It introduces 'adam', a framework that integrates Spark, Parquet, and Avro to improve bioinformatics tools and algorithms while emphasizing the need for bioinformaticians to adapt their software. The future of genomics is anticipated to involve larger datasets and increasingly complex analyses, necessitating efficient computational strategies.























































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)











