
Download as PDF, PPTX



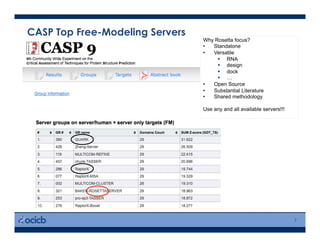
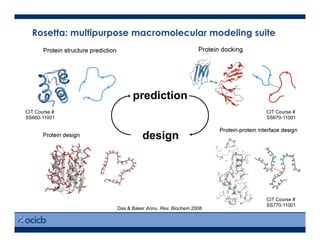
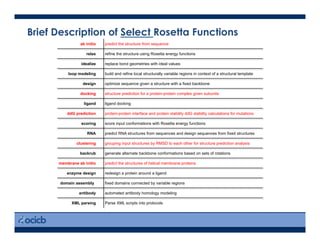
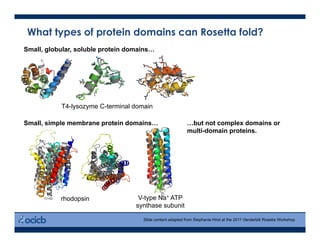
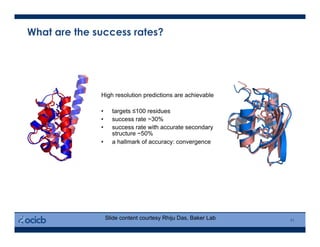
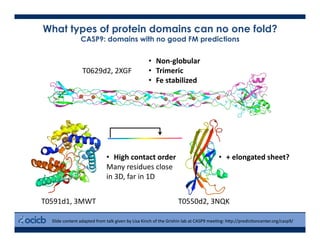
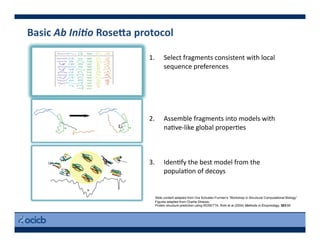
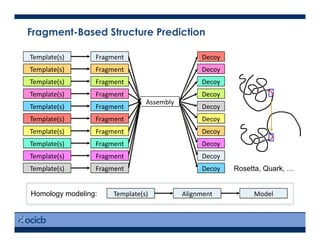

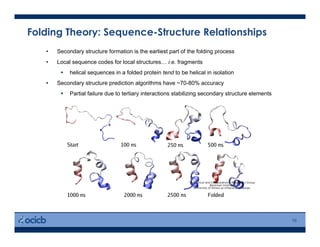


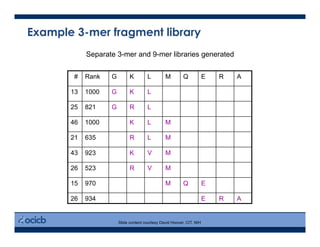
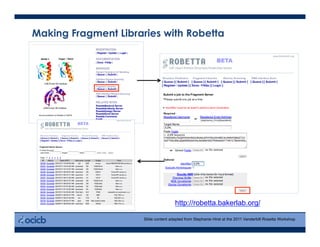


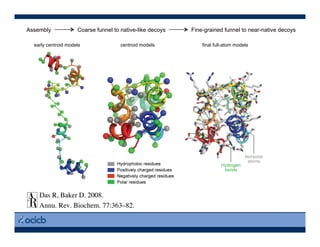

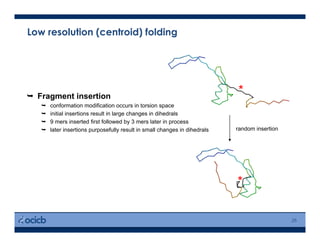

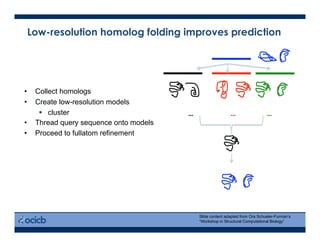
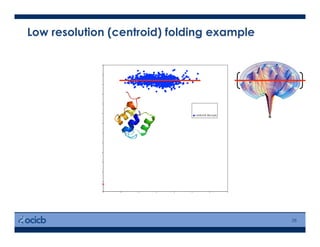
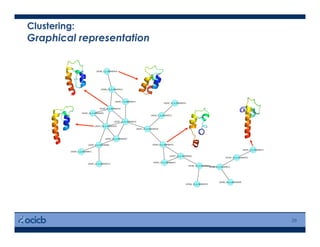
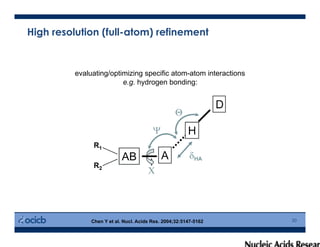

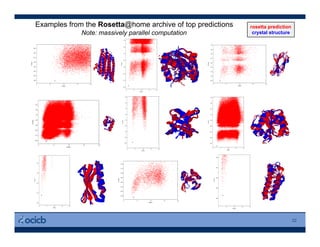
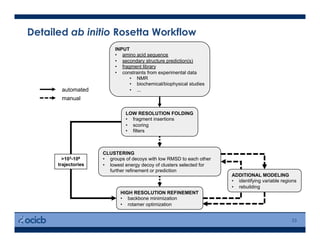
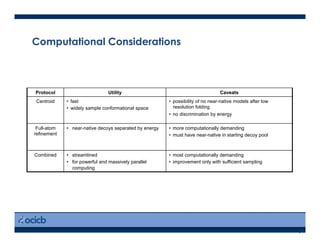
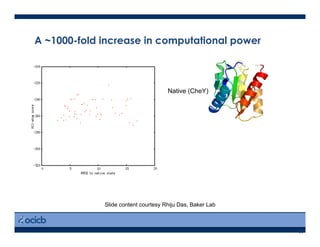
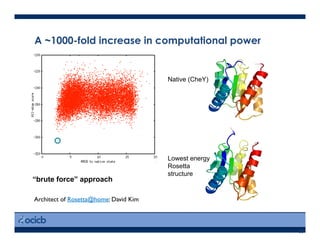
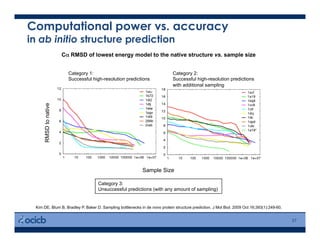
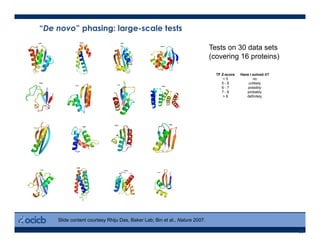
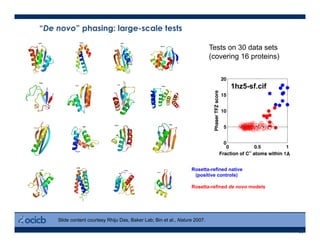
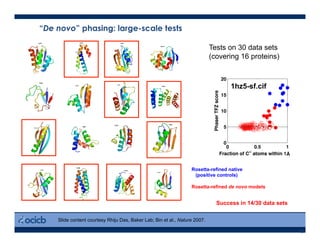
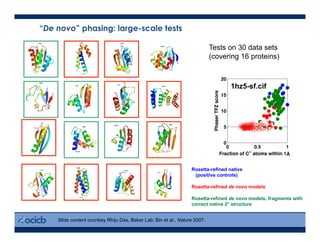


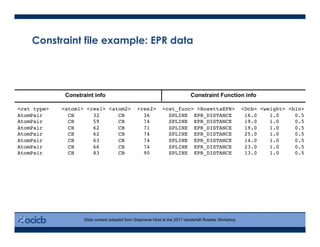



This presentation discusses protein structure prediction using Rosetta. It begins with an overview of the Critical Assessment of Protein Structure Prediction (CASP) experiments and notes that Rosetta is one of the top performing free-modeling servers. The presentation then describes the basic ab initio protocol used by Rosetta, which involves fragment insertion, scoring, and refinement. It also discusses limitations and success rates. Key aspects of the Rosetta energy functions and sampling algorithms are presented. Examples of specific Rosetta applications including low-resolution modeling and refinement are provided.












































![Following the Evolution of New Protein Folds via Protodomains [Report]](https://cdn.slidesharecdn.com/ss_thumbnails/blivencandidacyexam-130129194914-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)























![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)

















