Download as PDF, PPTX








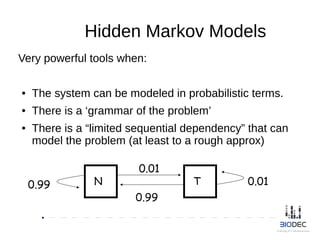















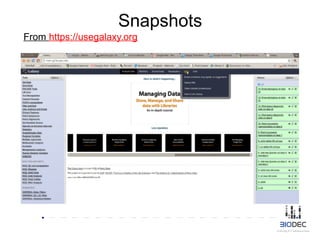
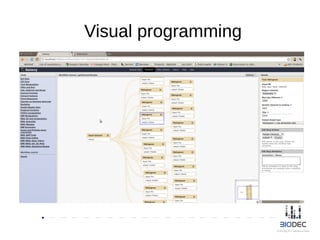




The document discusses the significance of Python in bioinformatics since 2002, highlighting its utility in bioinformatics software development, consulting, and creating annotated databases. It emphasizes the challenges of high-throughput data processing, the use of machine learning techniques, and the importance of tools like Biopython and Galaxy for efficient analysis and community collaboration. Key strategies for managing data quality, method selection, and community engagement in the bioinformatics field are also presented.















































