
Download as PDF, PPTX



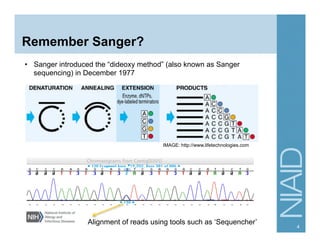

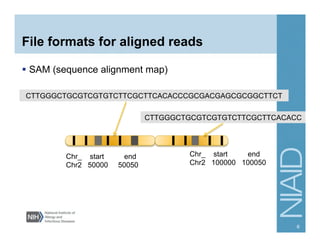


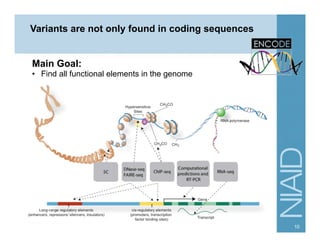
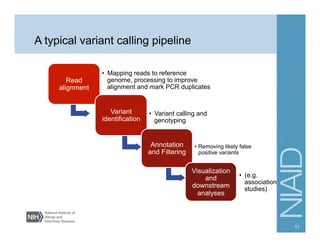

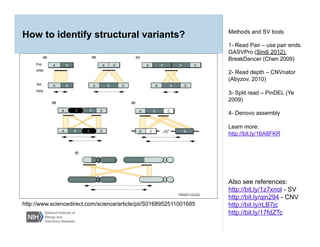
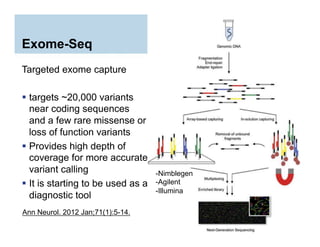

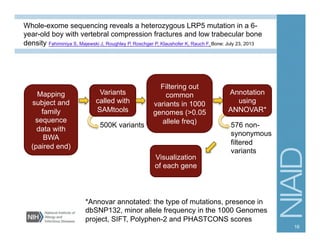
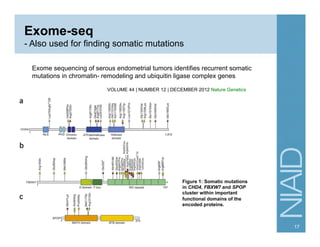






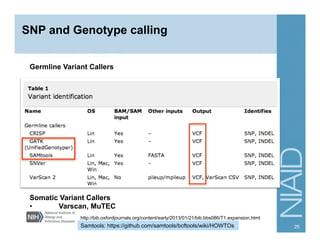
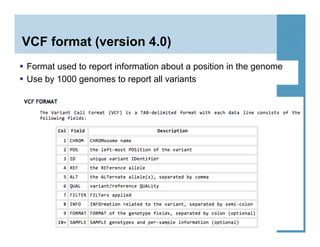

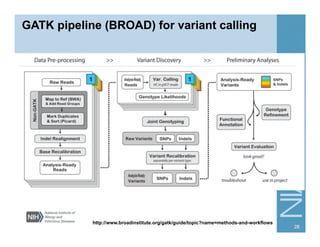

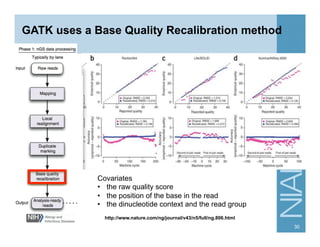


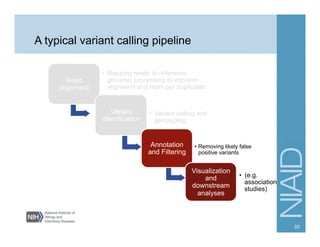
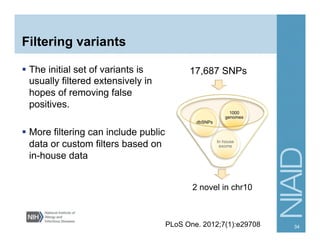



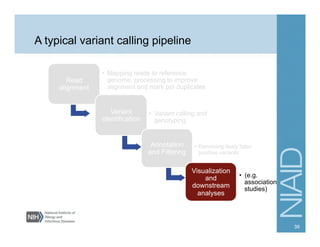

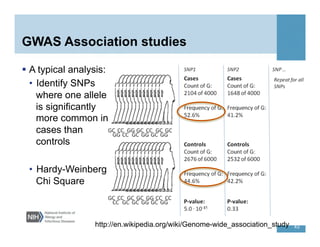




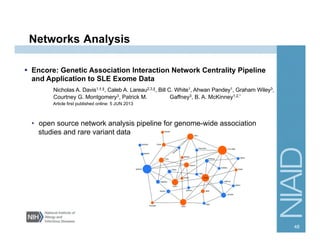

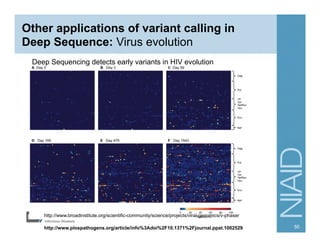
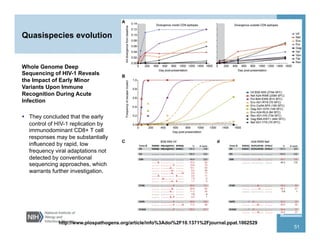
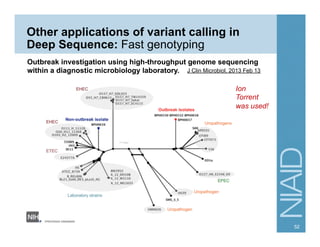


This document provides an overview of downstream analyses that can be performed after variant identification and filtering in a typical variant calling pipeline. It discusses visualization of variant data in each gene to identify potential causative variants. It also mentions association studies as another type of downstream analysis where variants are tested for association with disease phenotypes. The goal of downstream analyses is to help prioritize variants for further investigation.























































































