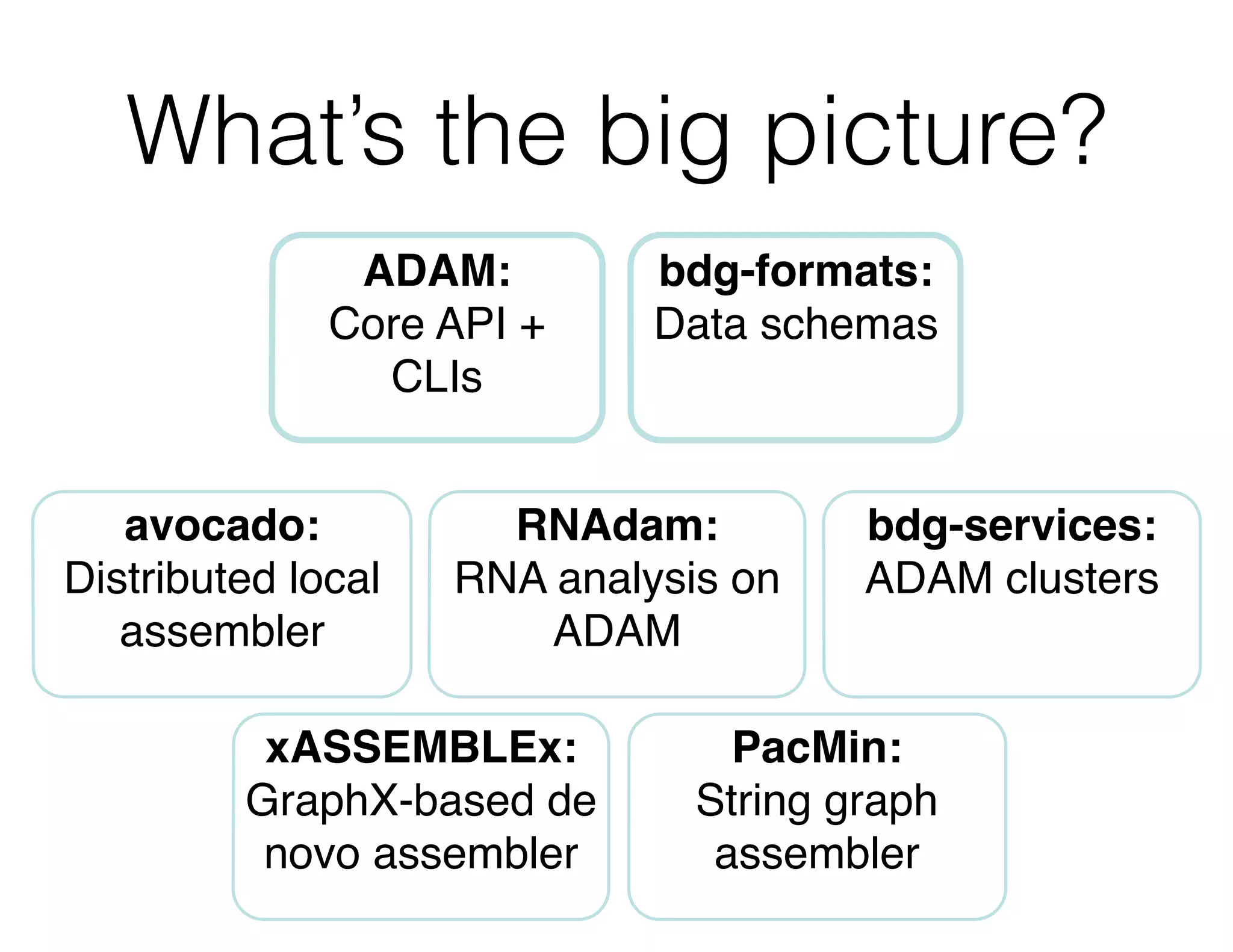
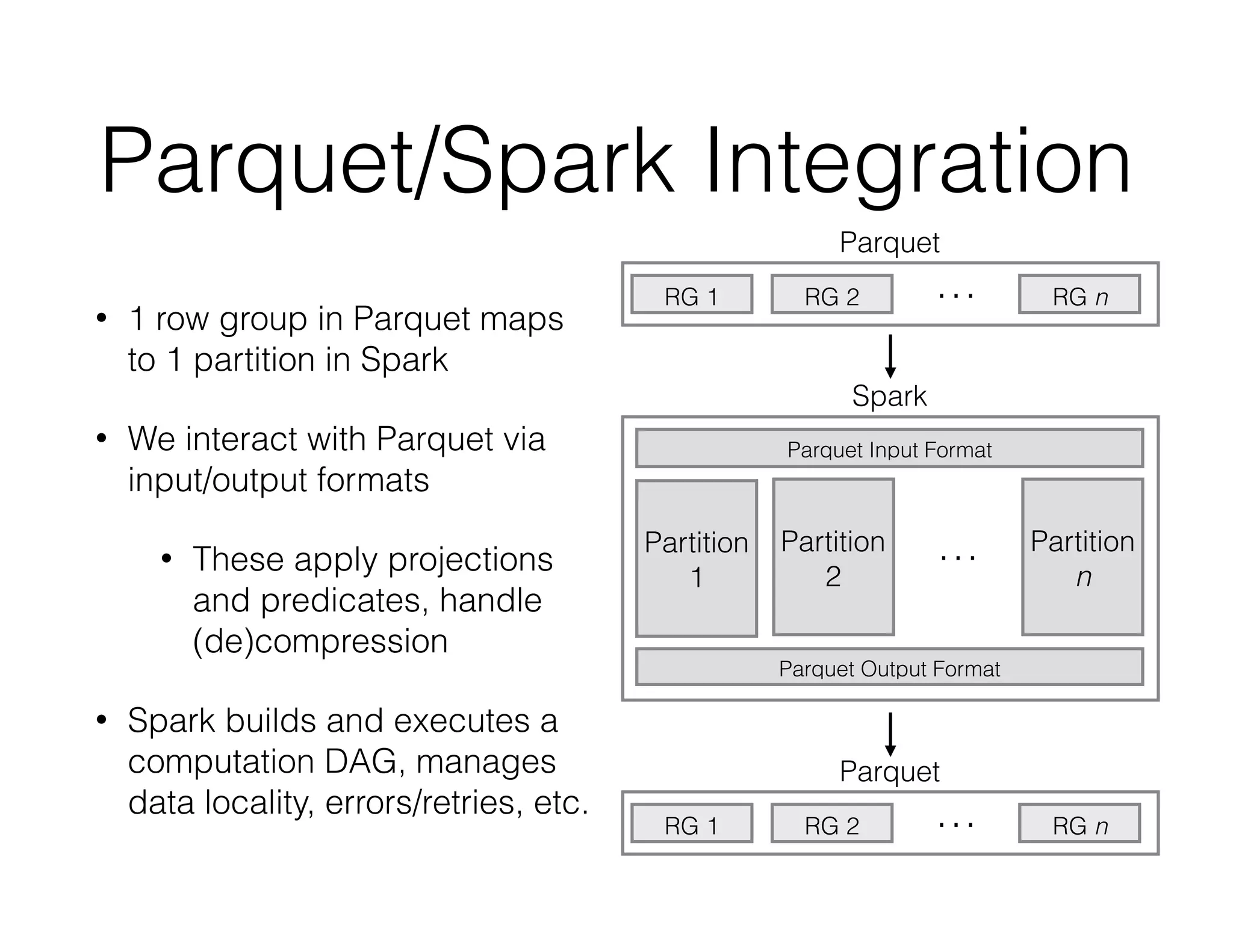
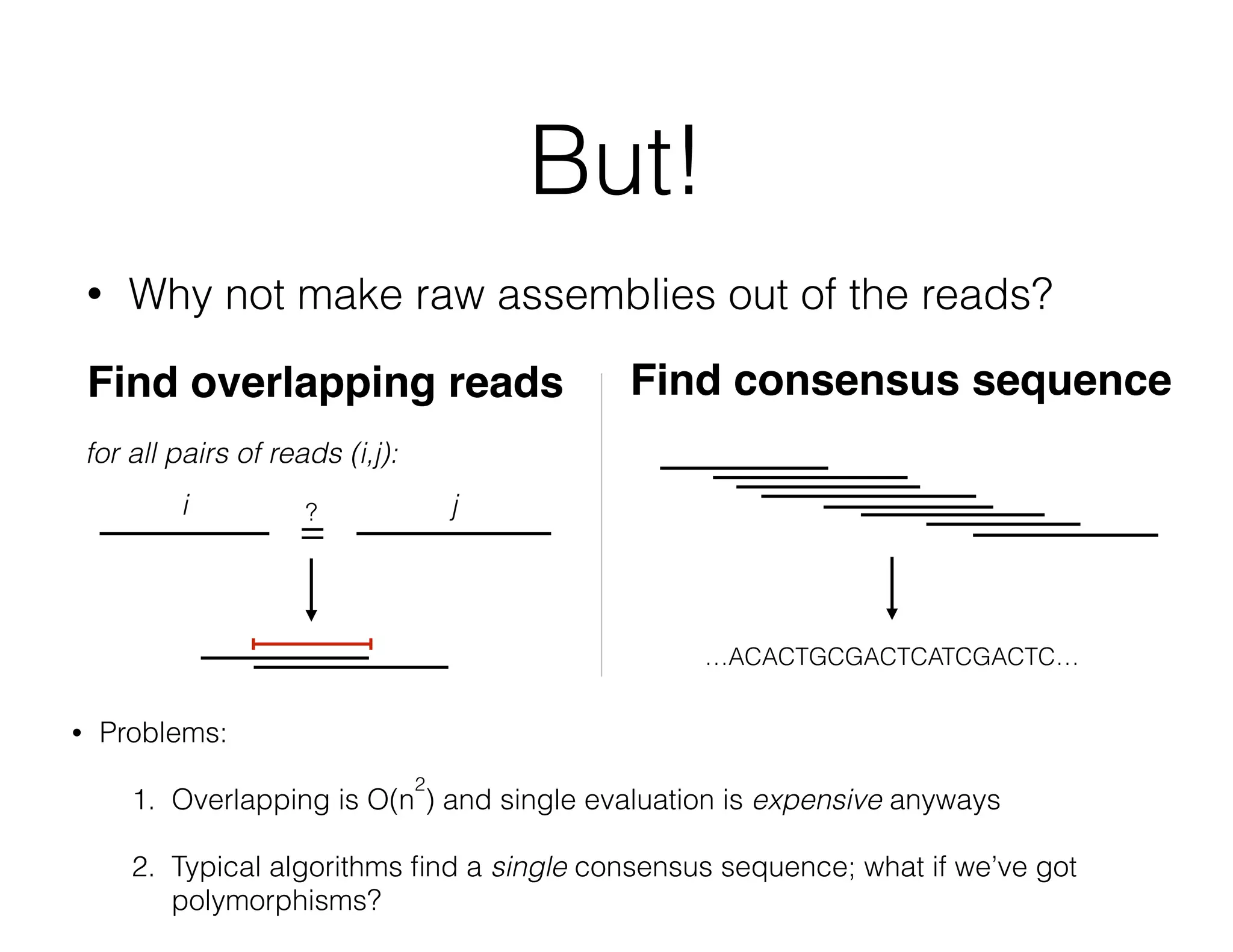
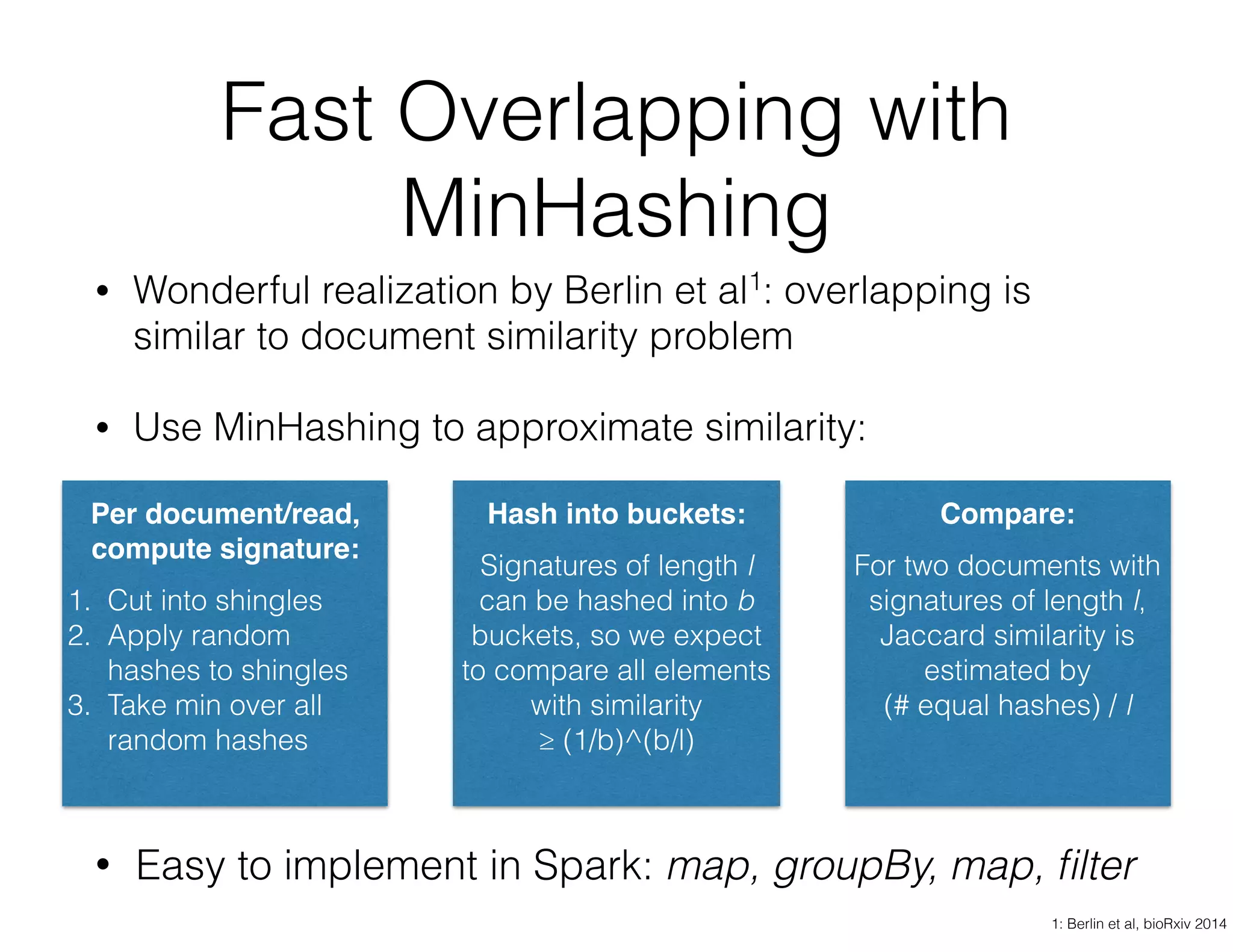
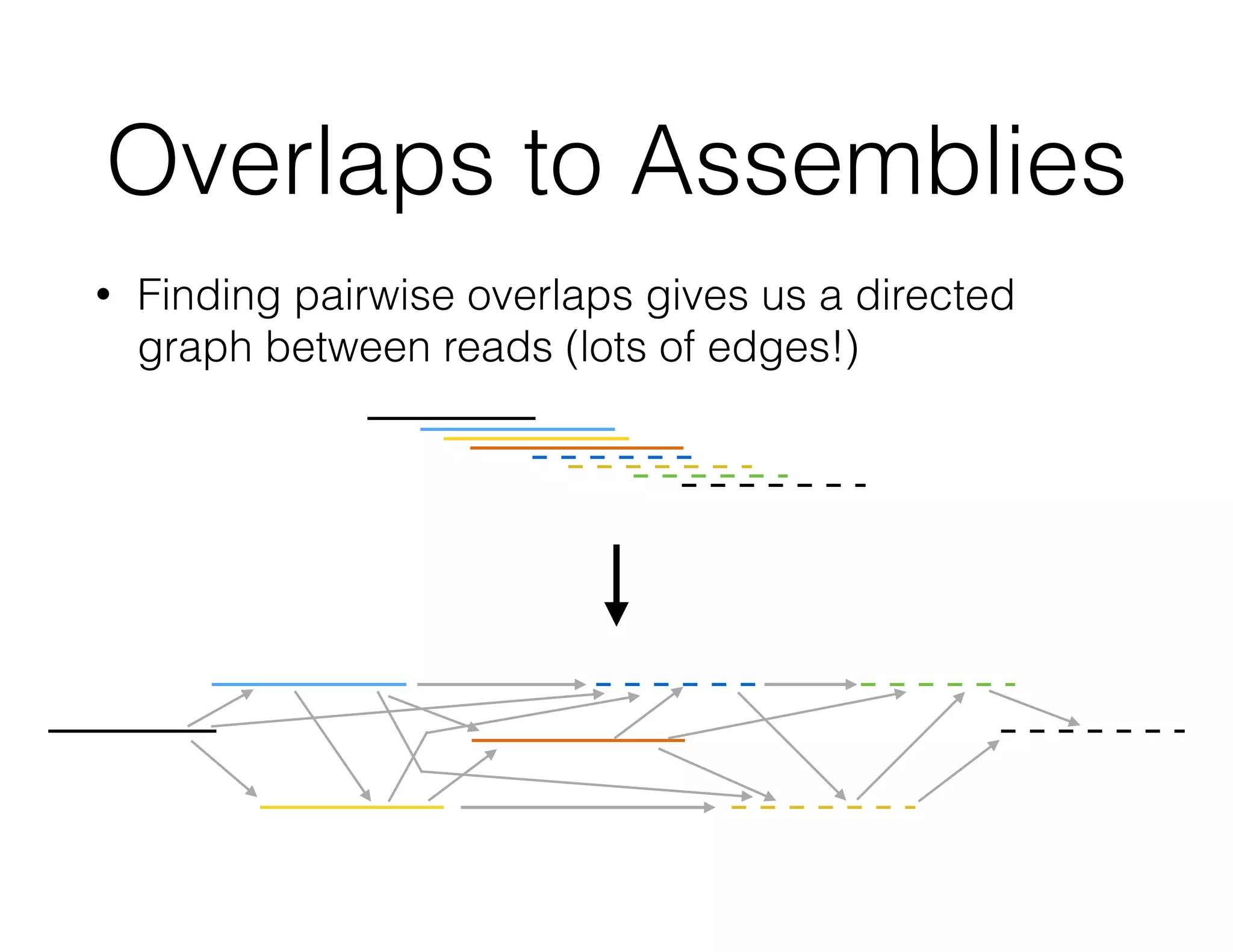
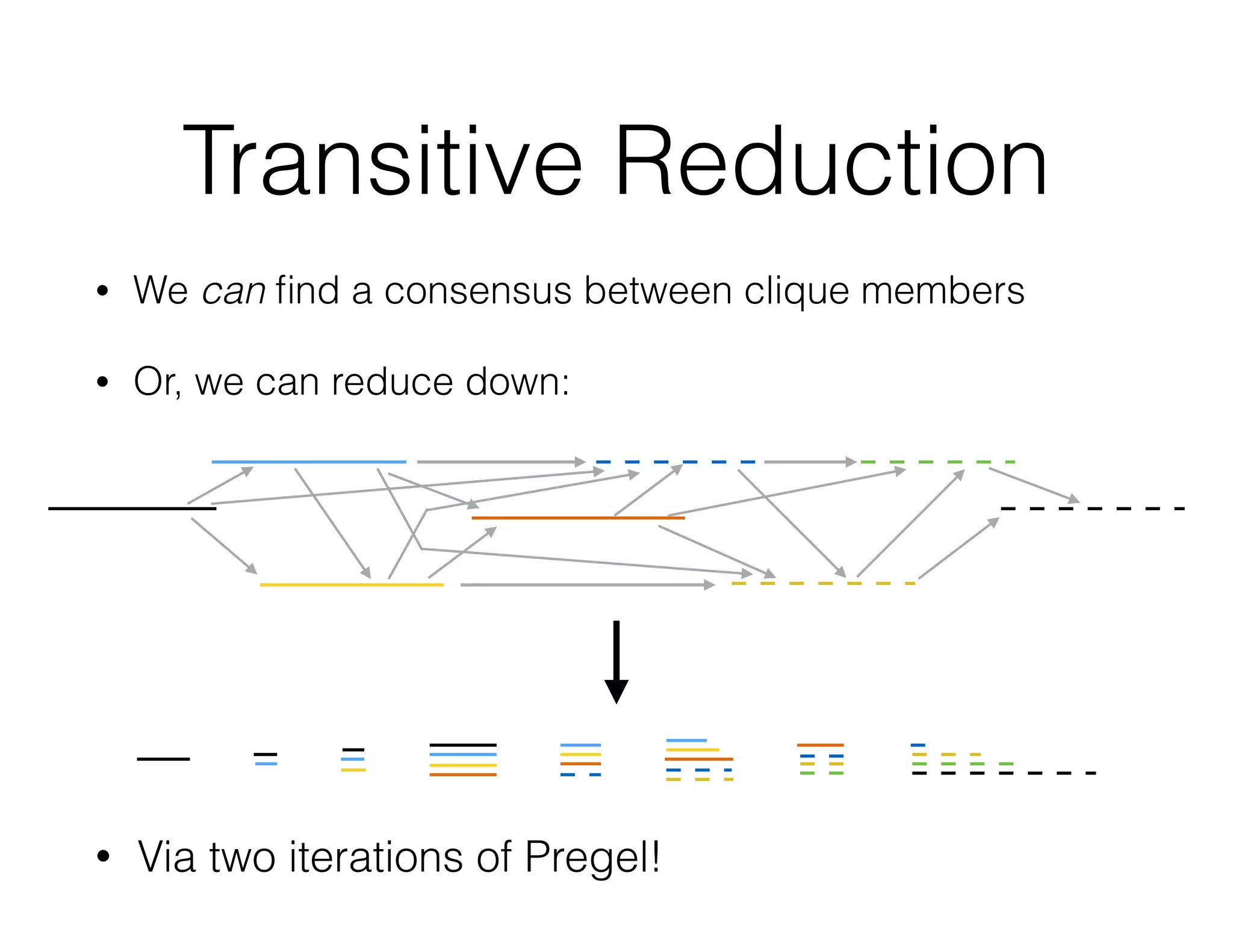
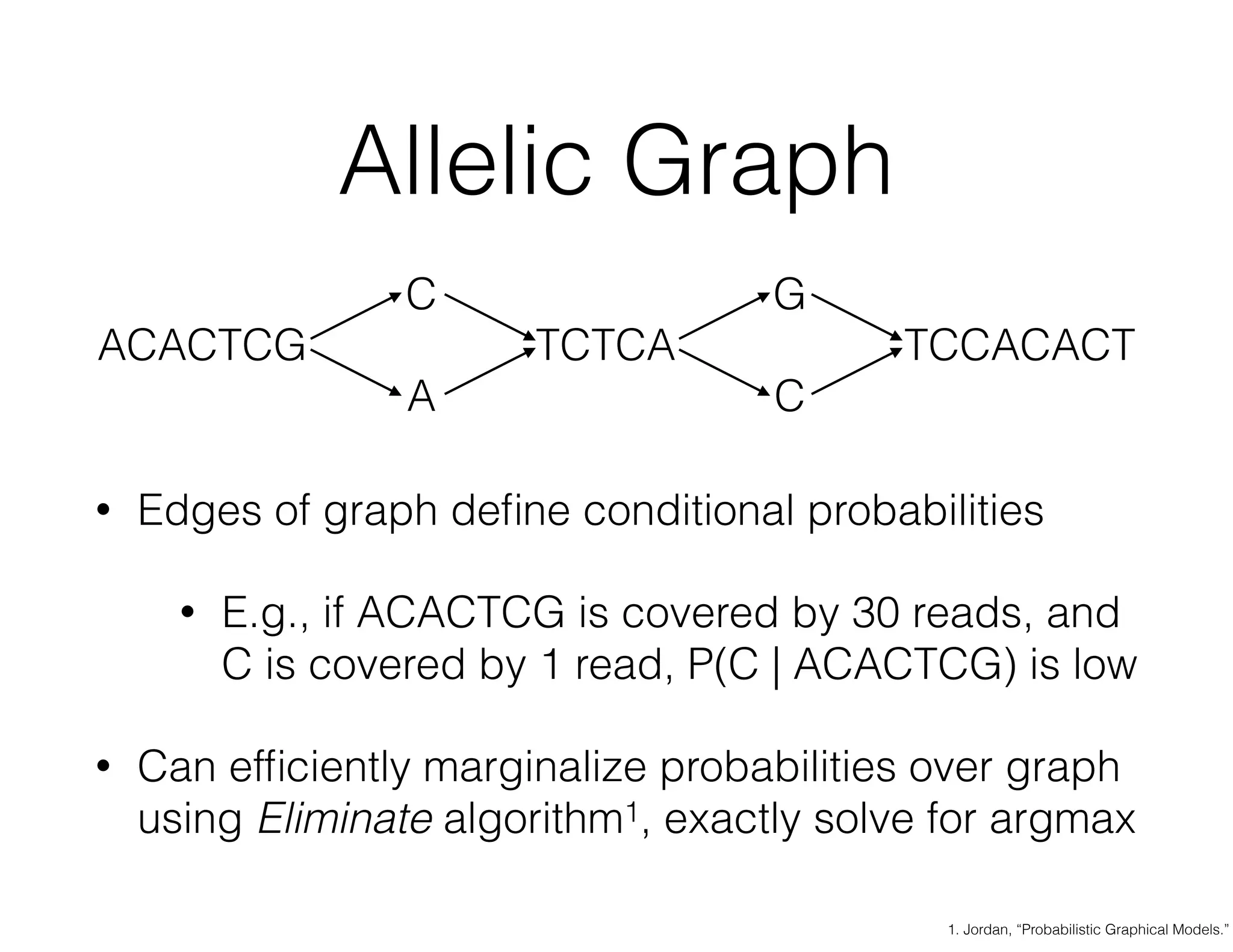
ADAM is an open source platform for scalable genomic analysis that defines a data schema, Scala API, and command line interface. It uses Apache Spark for efficient parallel and distributed processing of large genomic datasets stored in Parquet format. Key features of ADAM include its ability to perform iterative analysis on whole genome datasets while minimizing data movement through Spark. The document also describes using ADAM and PacMin for long read assembly through techniques like minhashing for fast read overlapping and building consensus sequences on read graphs.