Download as PDF, PPTX





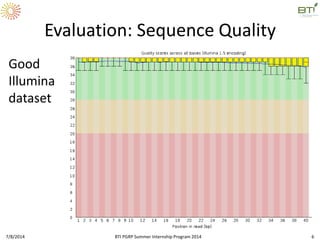
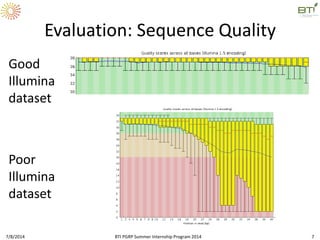
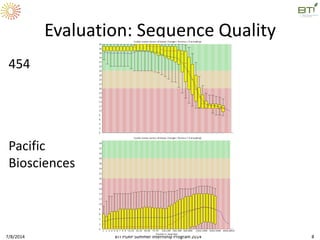
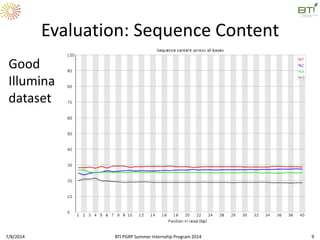
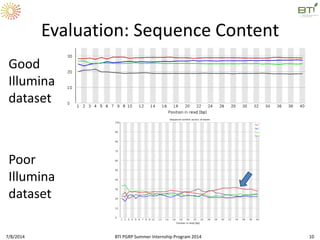
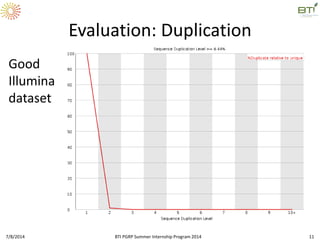
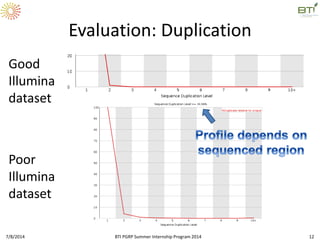


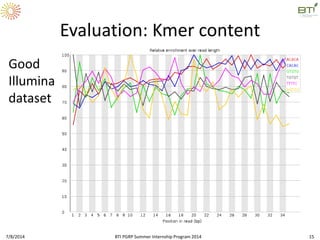
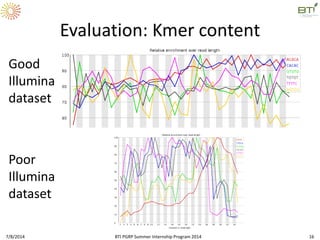
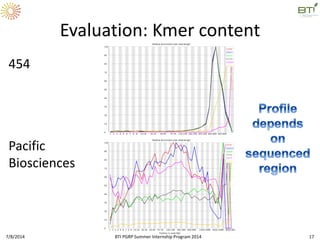





This document outlines exercises for quality control of NGS data from an Illumina sequencing experiment on tomato ripening stages. The exercises include: 1) evaluating raw fastq files for format and number of sequences; 2) using FastQC to analyze read quality scores, lengths, duplication levels, and k-mer content; and 3) preprocessing the reads using fastq-mcf to trim low quality ends and remove short reads before reanalyzing with FastQC. The goal is to learn how to evaluate NGS read quality and preprocess data prior to downstream analysis.























































































