
Downloaded 157 times






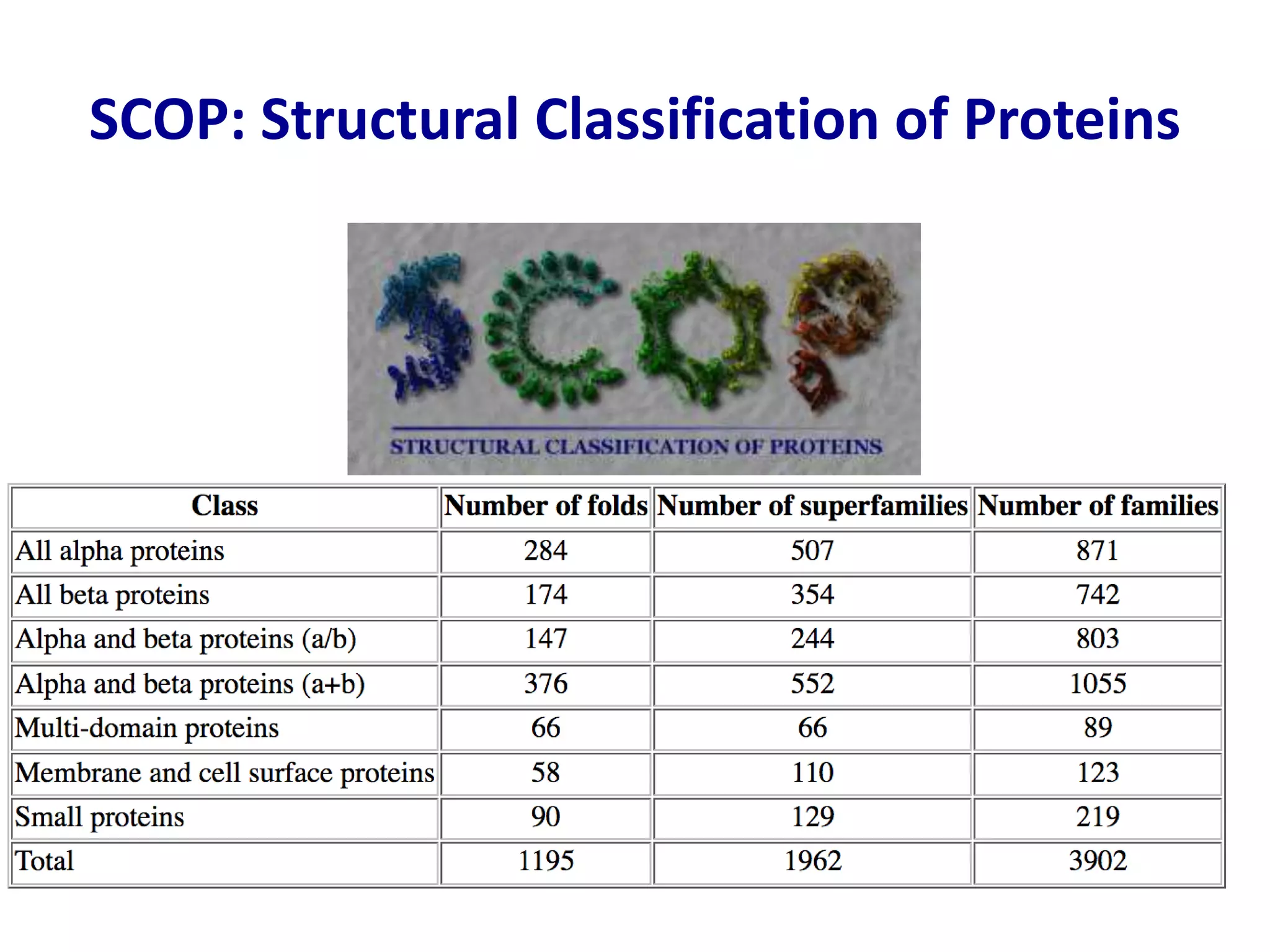
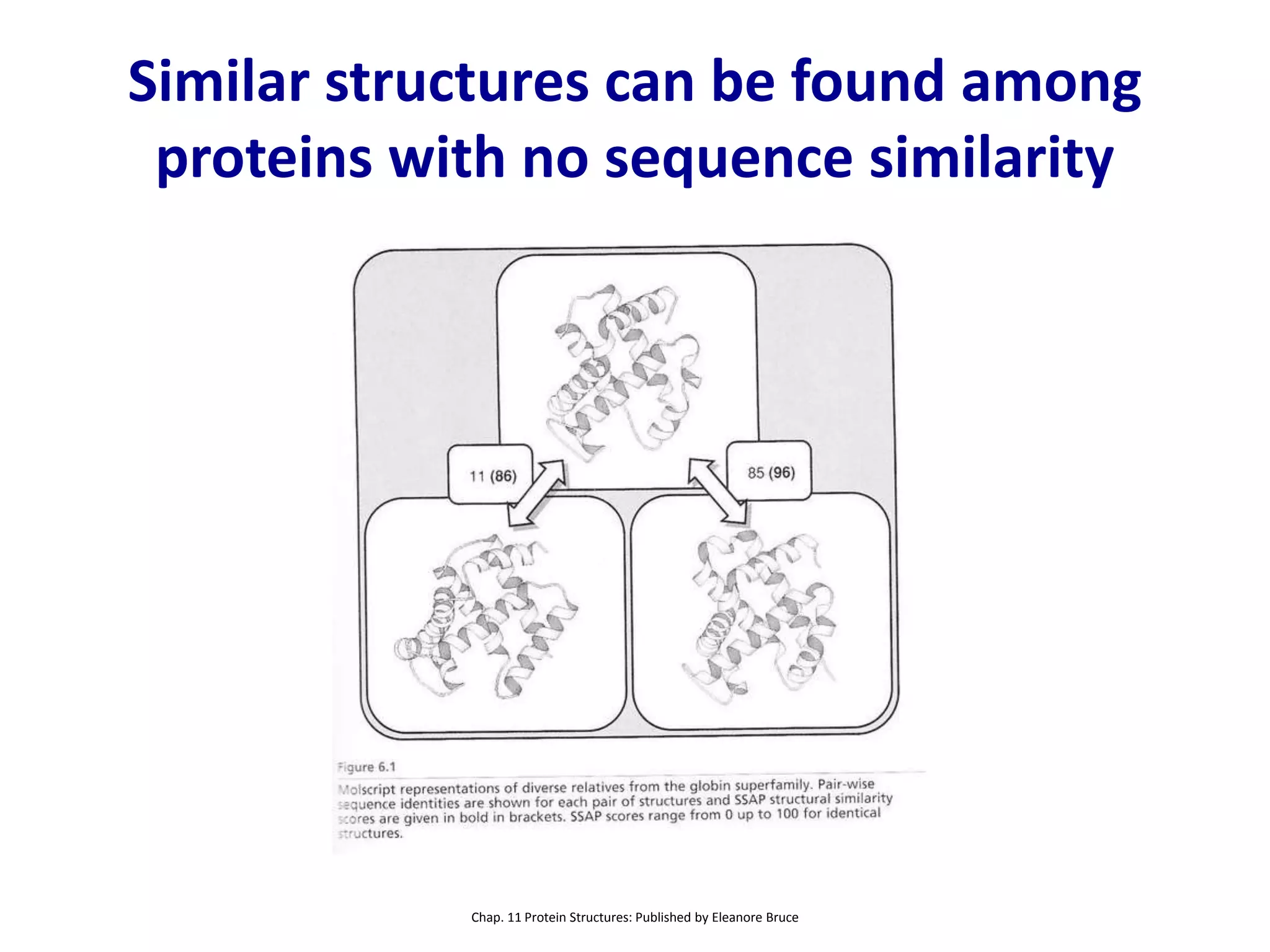

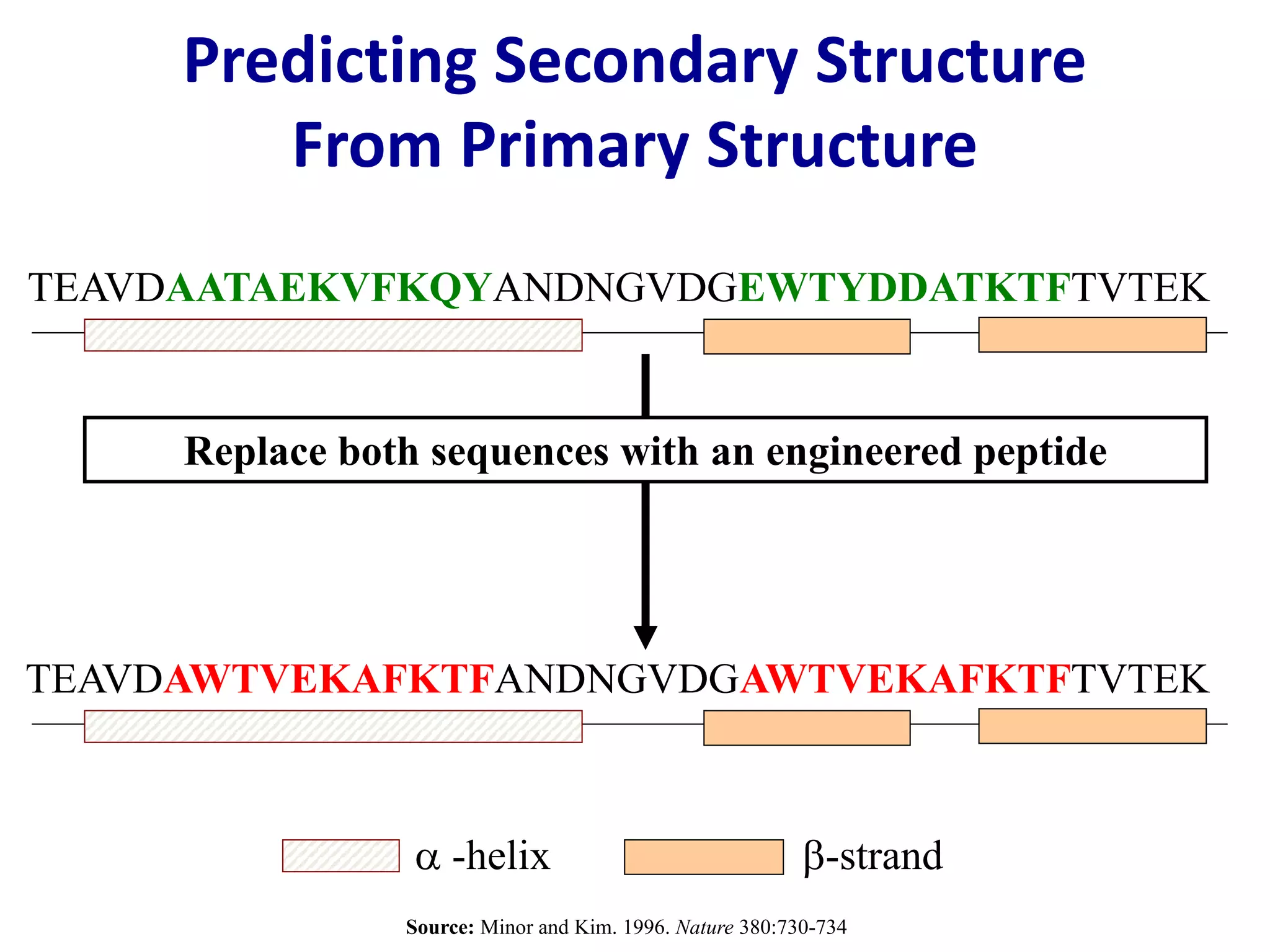

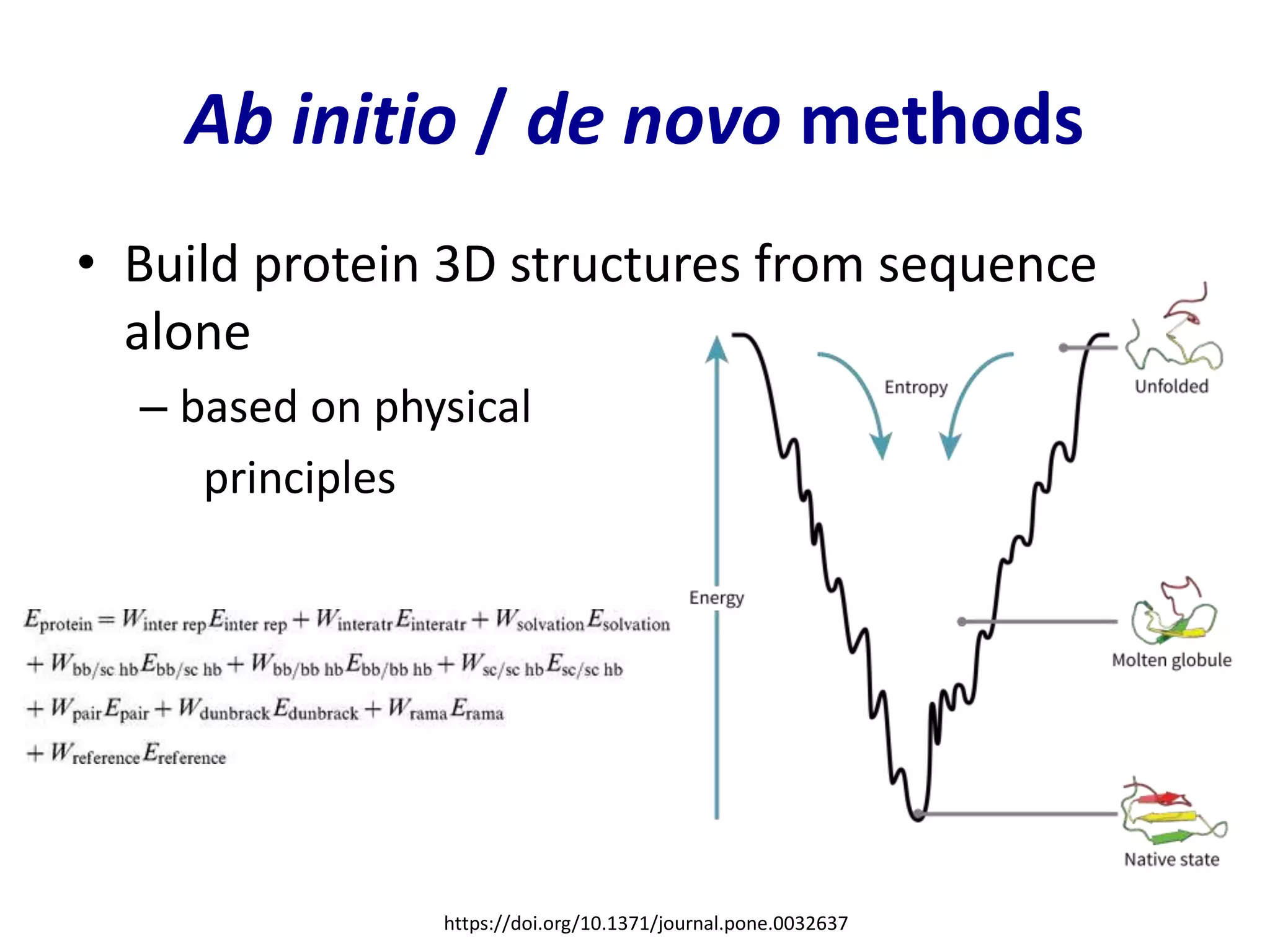
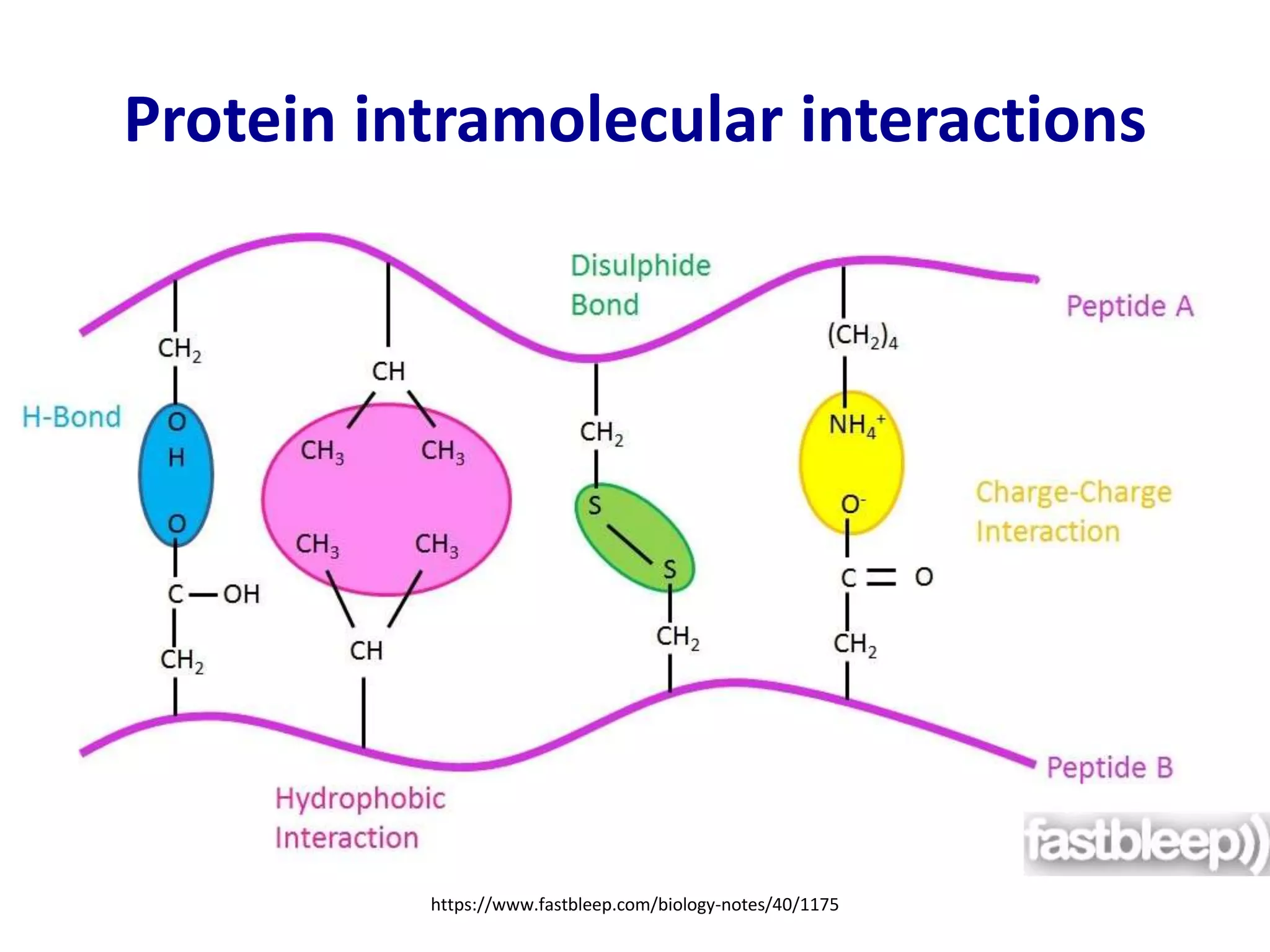

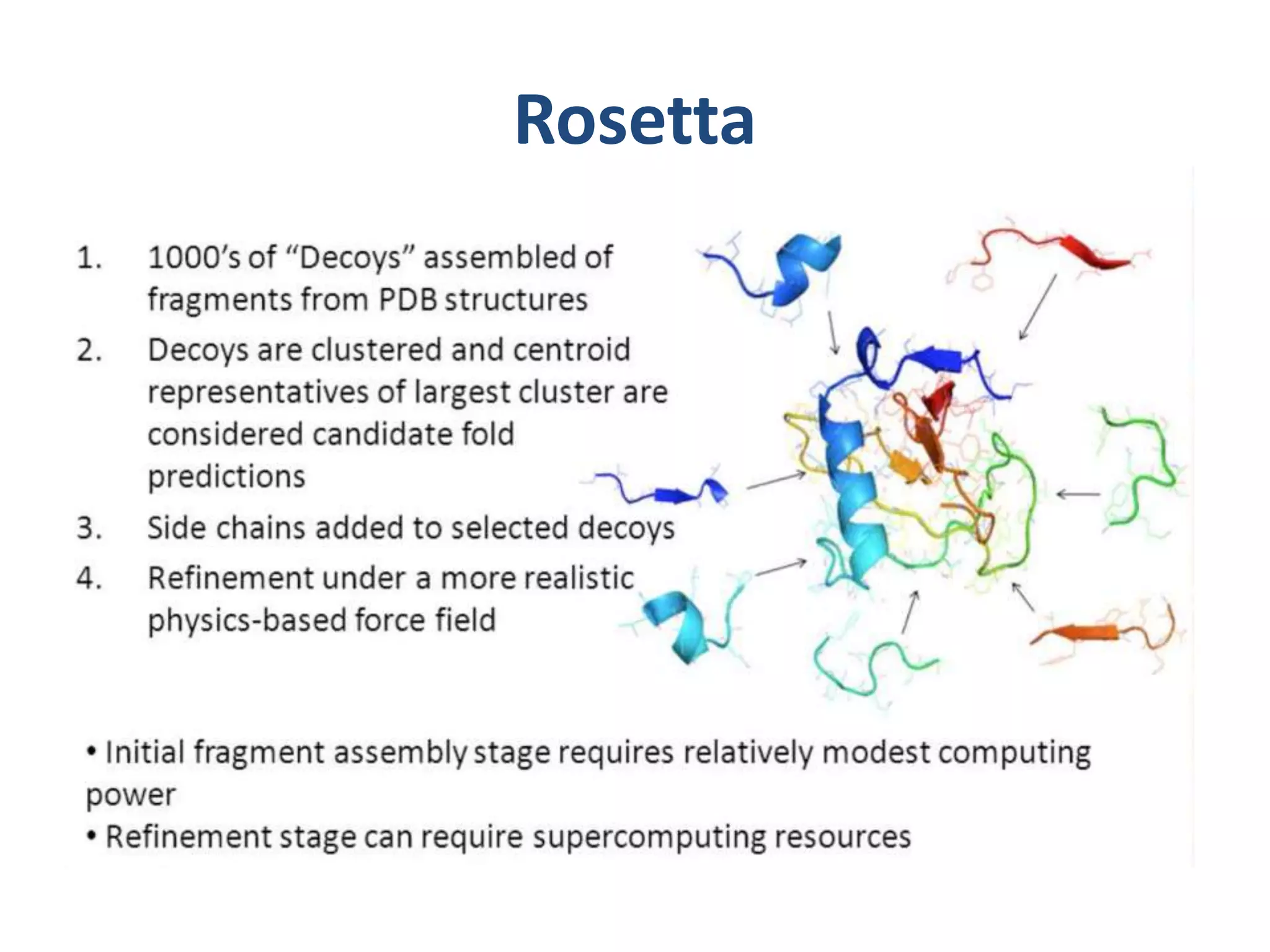
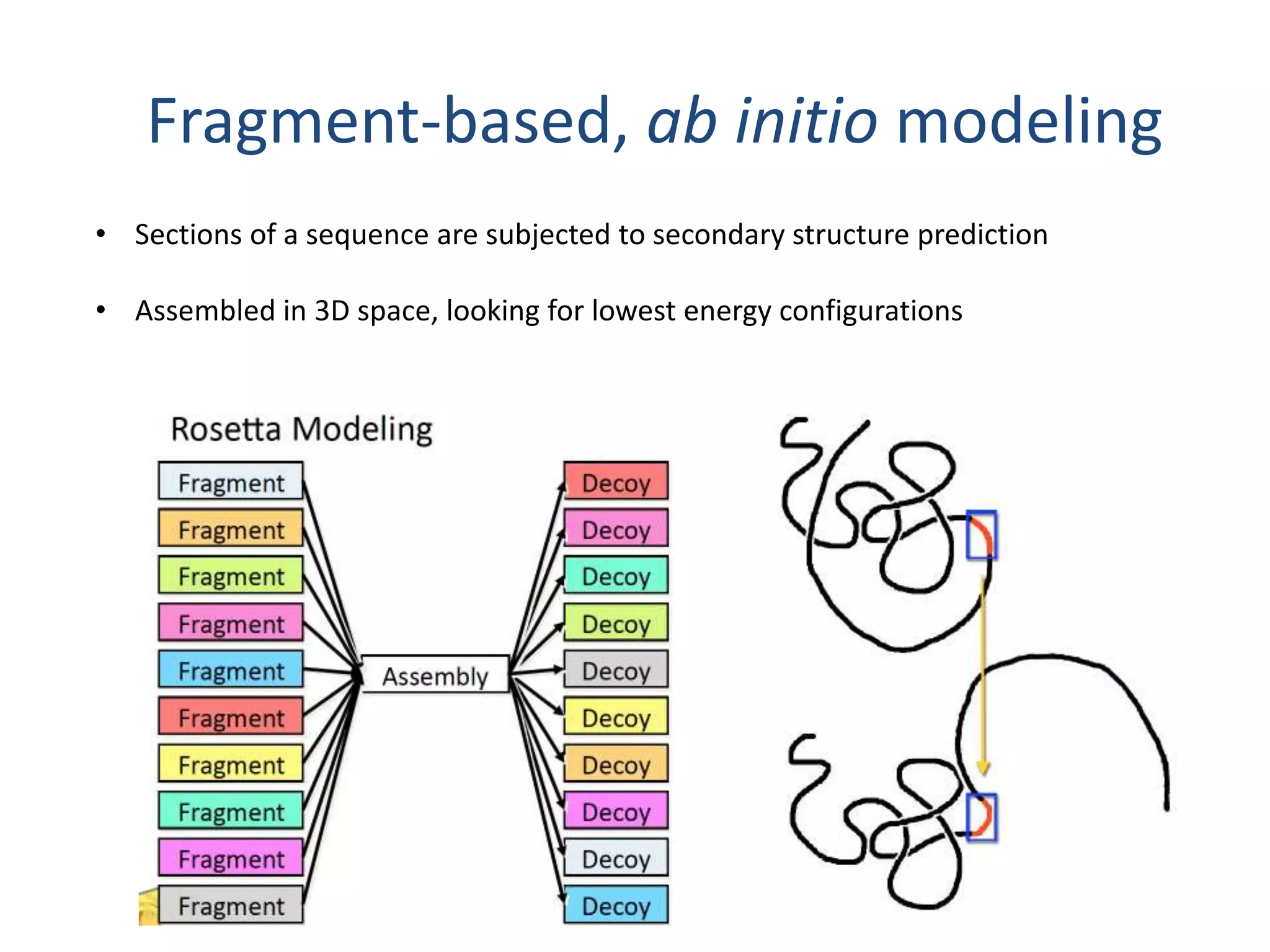



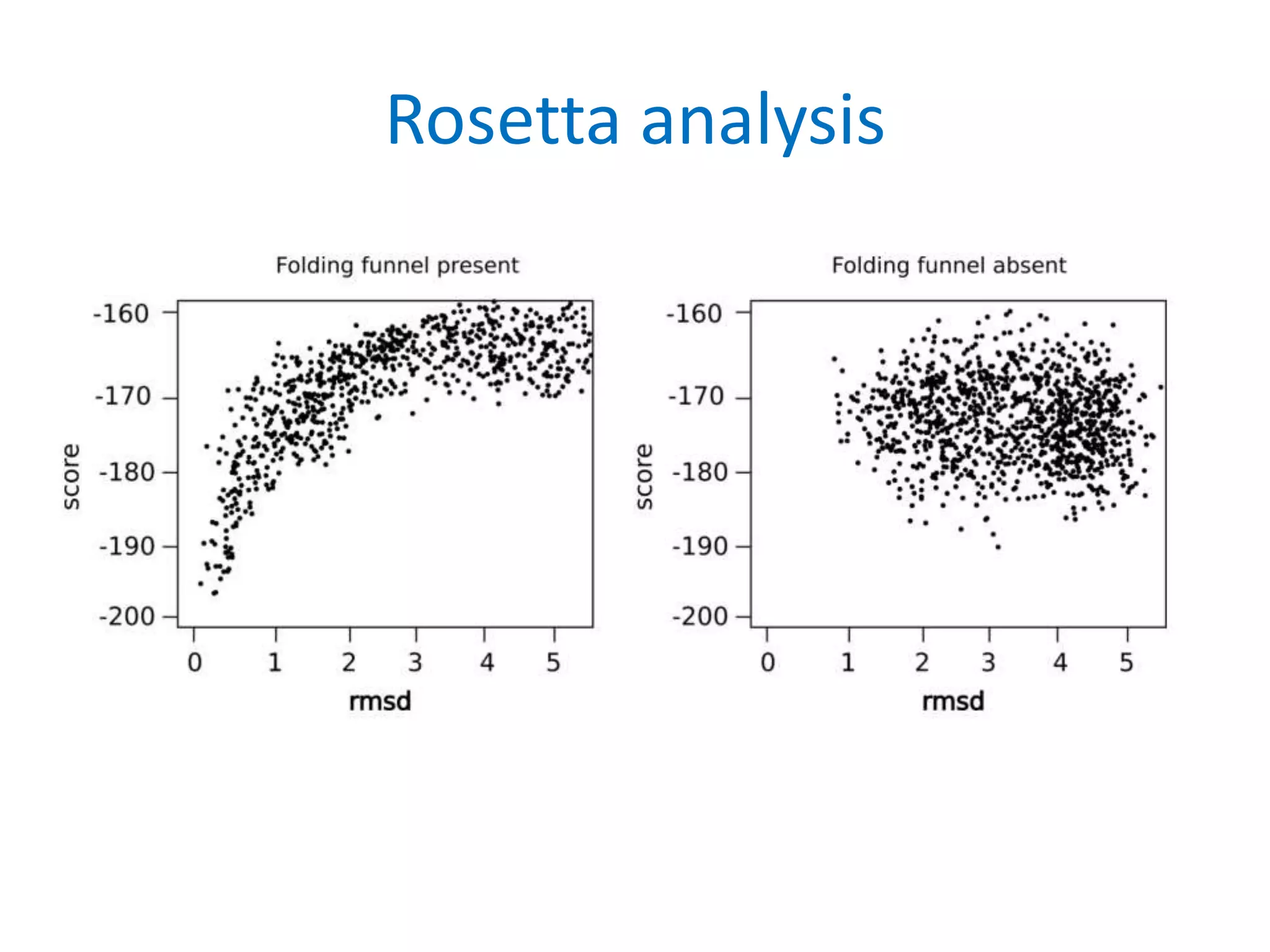
This document summarizes different computational methods for protein structure prediction, including homology modeling, fold recognition, threading, and ab initio modeling. Homology modeling relies on identifying proteins with similar sequences and known structures. Fold recognition and threading can be used when there are no homologs, to identify proteins with the same overall fold but different sequences. Ab initio modeling uses physics-based modeling and protein fragments to predict structure from sequence alone, and has challenges due to the vast number of possible conformations.























































































