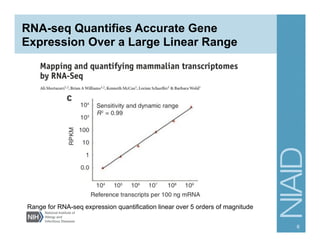
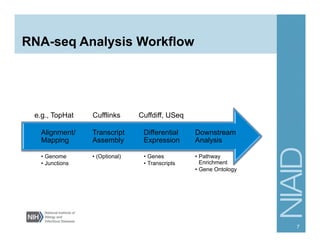
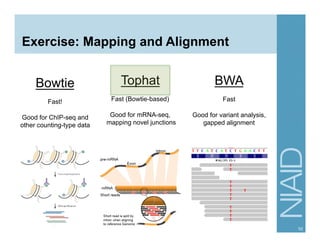
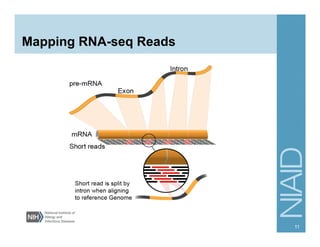
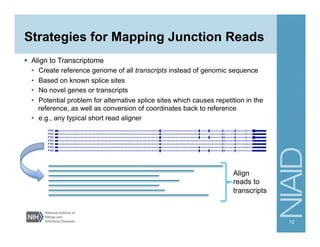
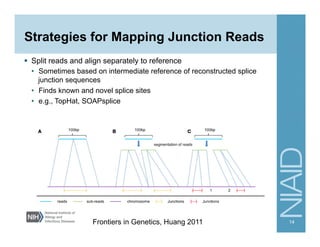
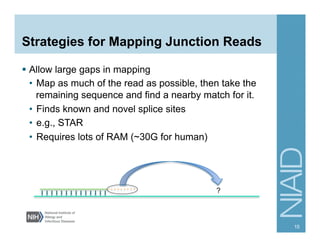
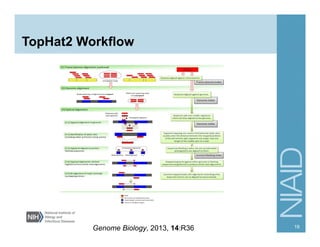
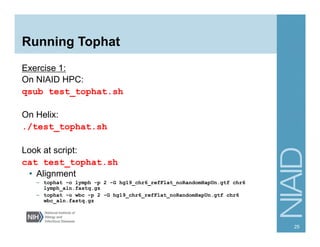
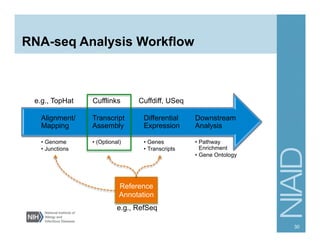
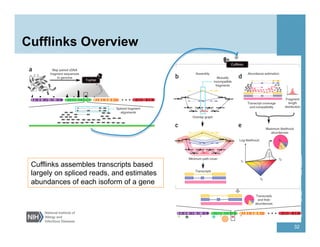
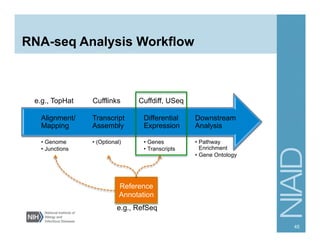
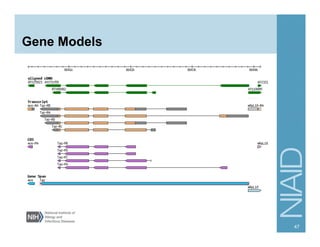
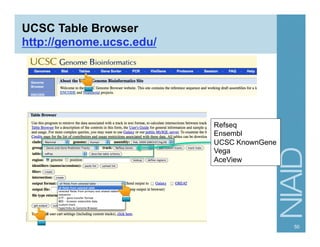
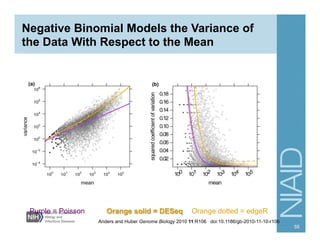
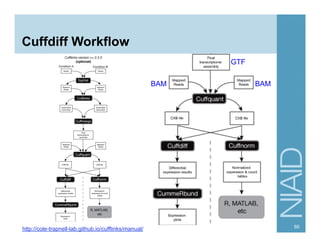
The document provides an overview of RNA sequencing (RNA-seq) analysis, including its advantages over traditional microarray methods, such as genome-wide coverage and reduced costs. It outlines the various steps in the RNA-seq workflow including read mapping with tools like Tophat2, transcript assembly with Cufflinks, and differential expression analysis. Additionally, the document describes library preparation protocols for different types of RNA and highlights the growing accessibility and cost-effectiveness of sequencing technologies.













![TopHat Command Line
22
tophat [options]* <index_base> <reads1_1> [reads1_2]
e.g., Paired-end
tophat hg19 SRR027894_1.fastq SRR027894_2.fastq
tophat hg19 SRR027894_1.fastq,SRR027895_1.fastq SRR027894_2.fastq,SRR027895_2.fastq
e.g., Single-end
tophat hg19 SRR036642.fastq
tophat hg19 SRR036642.fastq,SRR036643.fastq
Right mate in
paired-end
Single-end
or left mate in
paired-end
Index name
(genome)](https://image.slidesharecdn.com/rnaseq2015-02-18-170327193409-220906201705-2a94001a/85/rnaseq2015-02-18-170327193409-pdf-22-320.jpg)








![Cufflinks Options
cufflinks [options]* <aligned_reads.(sam/bam)>
Options:
-o output directory
-p number of threads/processors (default: 1)
-G <path> Use GTF/GFF annotation file to use determine isoform
expression. Do not assemble novel transcripts.
-g <path> Use GTF/GFF to guide assembly of annotated transcripts
(RABT); also assembly novel genes and isoforms
-M <path> GTF/GFF file containing regions to exclude from analysis, e.g.,
chrM, rRNA
-b <genome.fa> perform bias correction
-u multi-read correction calculation
--library-type <str> fr-unstranded (default), fr-firststrand (dUTP method), fr-
secondstrand (directional Illumina)
-F <0.0-1.0> minimum isoform fraction to include an isoform. (default: 0.1,
which means at least 10% of the most abundant isoform of the
gene)
Command:
cufflinks -o cuff_out -p 5 -g hg19_refFlat.gtf -M chrM_rRNA.gtf
-u -b genome.fa accepted_hits.bam
34
http://cufflinks.cbcb.umd.edu/manual.html](https://image.slidesharecdn.com/rnaseq2015-02-18-170327193409-220906201705-2a94001a/85/rnaseq2015-02-18-170327193409-pdf-34-320.jpg)





![Running Cuffcompare
cuffcompare [options] <transcripts1.gtf>
<transcripts2.gtf> …
e.g.,
cuffcompare -r hg19_chr6_refFlat_noRandomHapUn.gtf
lymph/transcripts.gtf wbc/transcripts.gtf
-r [file] Reference transcripts in gtf format
-R Ignore reference transcripts not found in
RNAseq sample
40](https://image.slidesharecdn.com/rnaseq2015-02-18-170327193409-220906201705-2a94001a/85/rnaseq2015-02-18-170327193409-pdf-40-320.jpg)















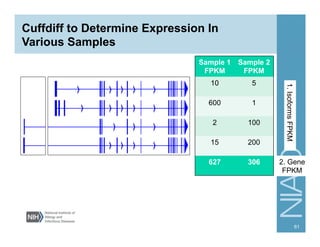
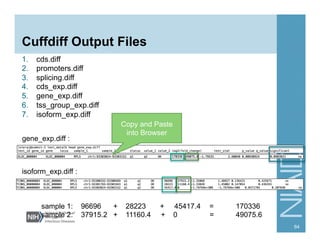
![Running Cuffdiff
cuffdiff [options] <transcripts.gtf> <sample1.bam>
<sample2.bam> …
e.g.,
cuffdiff -p 10 cuffcmp.combined.gtf lymph/
accepted_hits.bam wbc/accepted_hits.bam
-p [INT] Number of processors
-o Output directory (default = current dir)
-T Treat samples as a time series (default =
all against all comparison)
-u Multi-read correction for reads that map
to multiple places in the genome
Others (type “cuffdiff” to see other options)
62](https://image.slidesharecdn.com/rnaseq2015-02-18-170327193409-220906201705-2a94001a/85/rnaseq2015-02-18-170327193409-pdf-62-320.jpg)