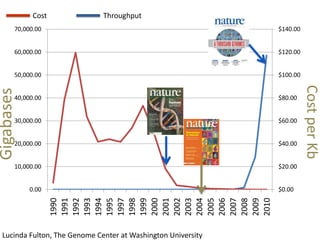
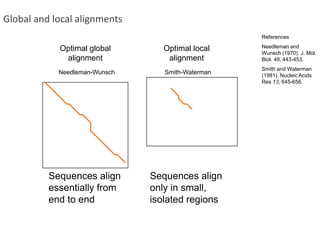
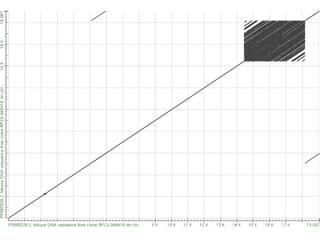
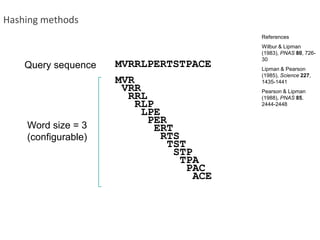
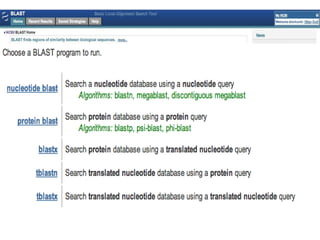
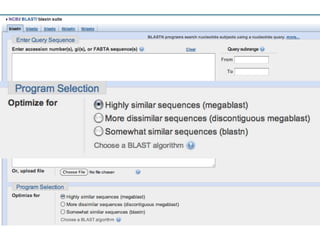
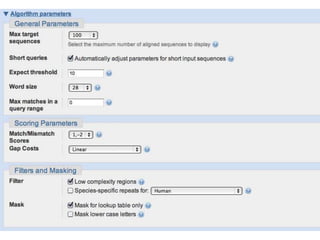
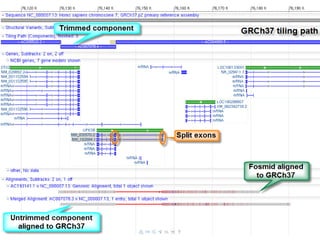
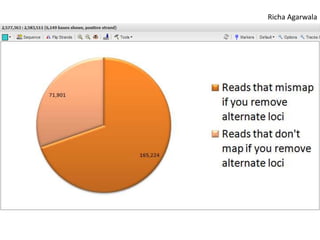
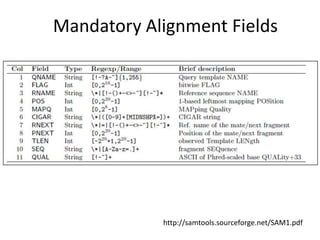
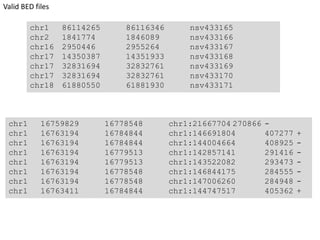
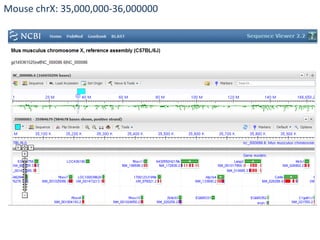
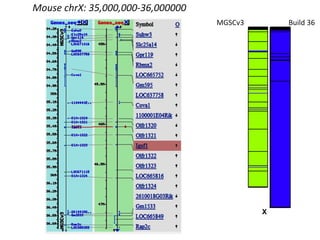
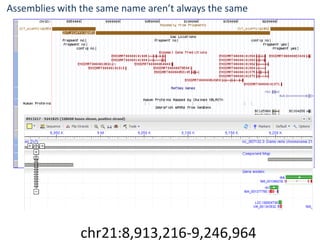
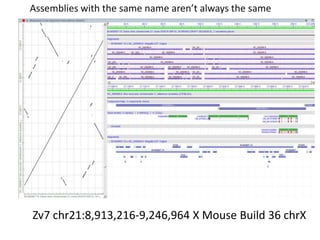
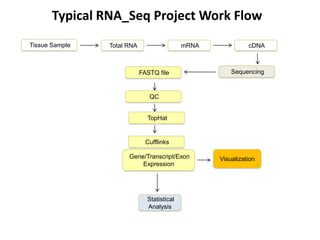
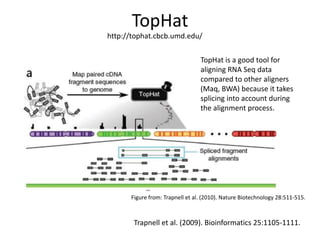
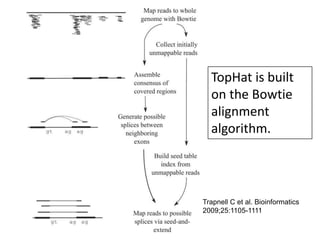
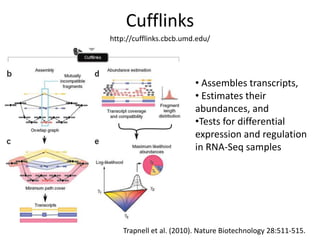
The document summarizes an IMGS 2011 bioinformatics workshop. It discusses next-generation sequencing technologies including Roche 454, Illumina/Solexa, and AB SOLiD. It also covers topics like sequence alignments, file formats, tools for analysis including BWA and TopHat, and visualization. The document provides links to video tutorials and resources on sequencing technologies, alignments, and analyzing RNA-seq data.