Downloaded 538 times

















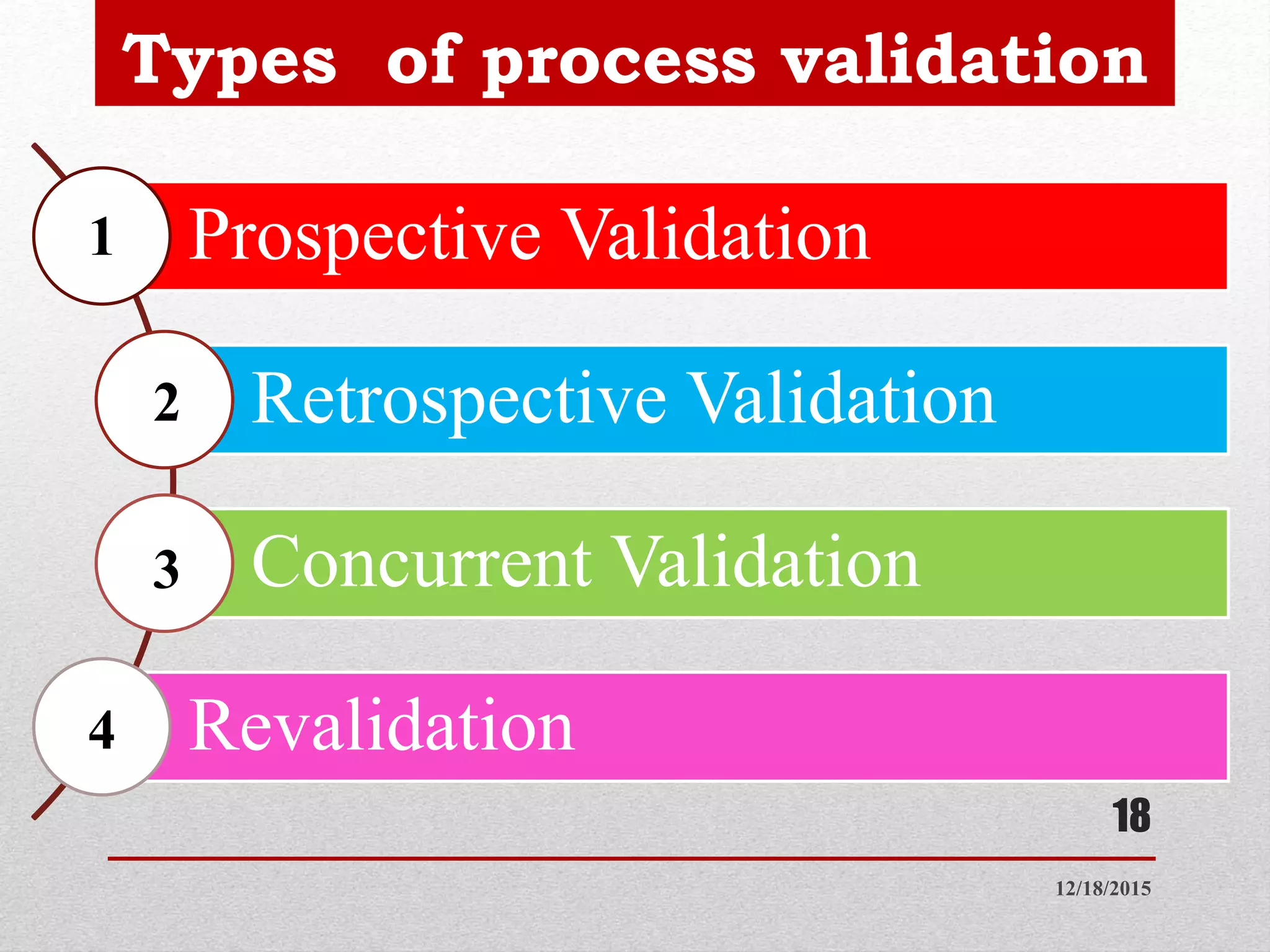

























Process validation is establishing documented evidence that a process will consistently produce a product meeting predetermined specifications. This presentation discusses process validation, including its definition, scope, objectives, types (prospective, retrospective, concurrent, revalidation), stages, responsibilities of different departments, protocols, sampling procedures, acceptance criteria, and reports. Key aspects of process validation include protocols, sampling plans, specifications, batch execution records, and data analysis to ensure a process is capable of reproducible commercial manufacturing of pharmaceutical products that meet quality standards.












































































