Downloaded 62 times







![CLASSIFICATION
A] Primary: Progressive muscle dystrophy
B] Secondary:
1] Inflammatory: polymyositis
2] Metabolic: Periodic familial paralysis
3] Endocrinal: thyrotoxic .Chushing.
4] Drug induced: corticosteroid. Statin .
5] Miscellaneous: carcinomatous](https://image.slidesharecdn.com/myopathyundergrad-160701160220/85/Myopathy-undergraduate-8-320.jpg)



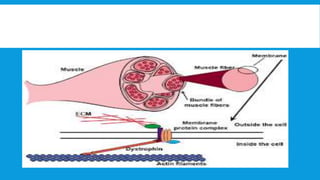

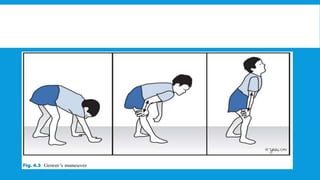


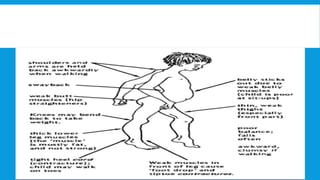


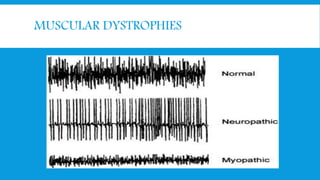








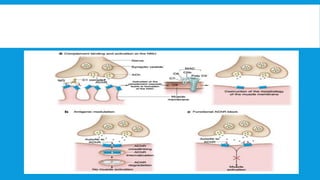


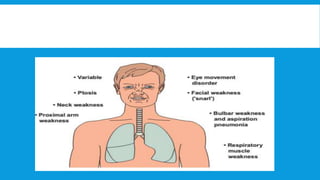

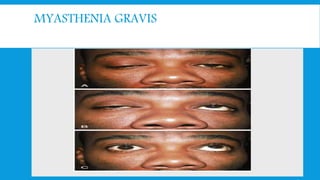
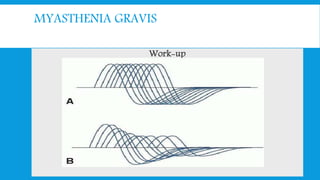






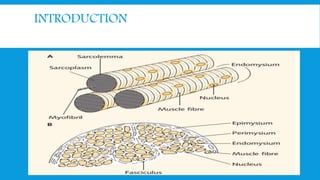
Skeletal muscle disorders can be classified as either primary muscle diseases or secondary disorders caused by other conditions like inflammation, metabolic abnormalities, or drugs. Progressive muscle dystrophies are a primary cause and include Duchenne muscular dystrophy and Becker muscular dystrophy, which are caused by mutations in the dystrophin gene. Symptoms include weakness, wasting, and pseudohypertrophy. Management focuses on rehabilitation, steroids, respiratory support, and future gene therapies. Myasthenia gravis is an autoimmune disorder where antibodies target acetylcholine receptors, causing fluctuating weakness. Diagnosis involves the Tensilon test and repetitive nerve stimulation with treatment consisting of cholinesterase inhibitors, steroids, plasma exchange,































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)










