Downloaded 752 times
![Neuromuscular Disorders –
Duchenne and Becker’s Muscular
Dystrophy
Dr. Kalpana Malla
MD Pediatrics
Manipal Teaching Hospital
Download more documents and slide shows on The Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/ms-duchenneandbeckers-120108092843-phpapp02/85/Muscular-Dystrophy-Duchenne-and-Becker-s-1-320.jpg)
![Neuromuscular Disorders –
Duchenne and Becker’s Muscular
Dystrophy
Dr. Kalpana Malla
MD Pediatrics
Manipal Teaching Hospital
Download more documents and slide shows on The Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/ms-duchenneandbeckers-120108092843-phpapp02/75/Muscular-Dystrophy-Duchenne-and-Becker-s-1-2048.jpg)
























































![Thank you
Download more documents and slide shows on The
Medical Post [ www.themedicalpost.net ]](https://image.slidesharecdn.com/ms-duchenneandbeckers-120108092843-phpapp02/85/Muscular-Dystrophy-Duchenne-and-Becker-s-64-320.jpg)

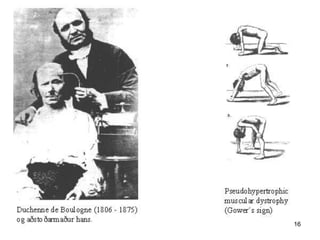
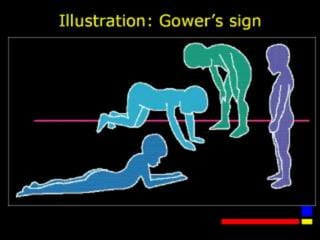
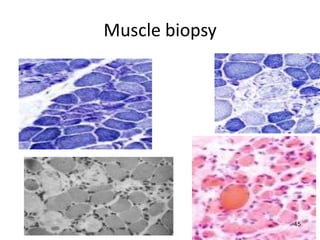
Duchenne muscular dystrophy is a genetic disorder characterized by progressive muscle weakness. It is caused by mutations in the gene encoding dystrophin, and mainly affects boys. Clinical features include difficulty walking, calf pseudohypertrophy, loss of ambulation by age 12, wheelchair dependence, scoliosis, and death often by age 18 from respiratory or cardiac failure. Diagnosis involves elevated creatine kinase levels, muscle biopsy showing dystrophin deficiency, and genetic testing. There is no cure, but management focuses on maintaining mobility and function.











































































































