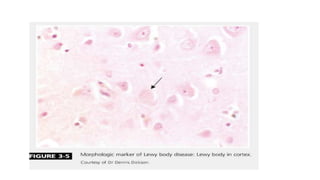
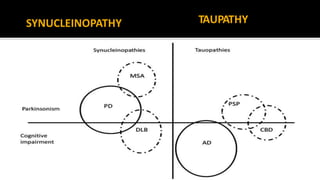
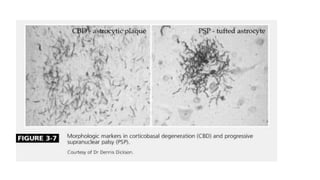
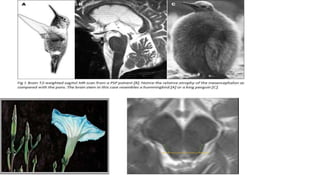
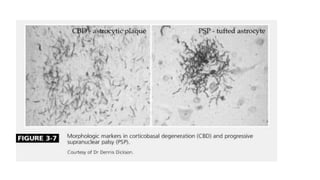
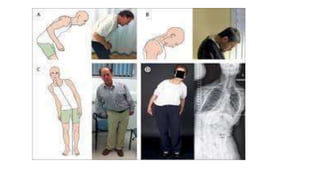
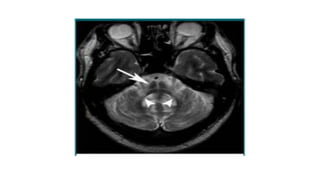
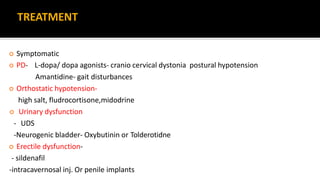
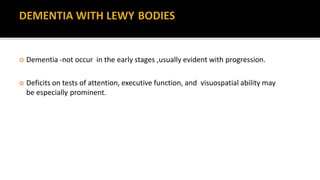
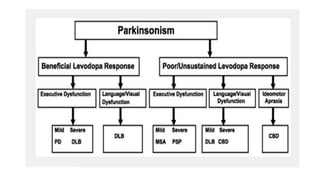
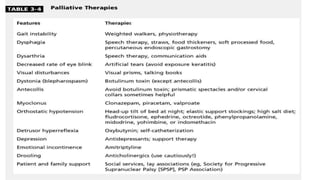
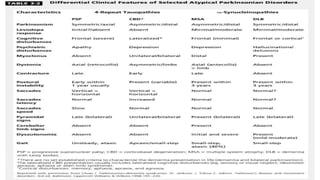
This document discusses progressive supranuclear palsy (PSP). It begins by classifying PSP and outlining its diagnostic criteria and phenotypes. Key signs of PSP include early vertical supranuclear gaze palsy, postural instability with falls, and poor response to levodopa. Investigations like MRI, PET, and CSF analysis can aid in diagnosis. Pathology involves tau neurofibrillary tangles in brainstem nuclei. There is no effective treatment, only supportive and symptomatic measures. The document also briefly discusses multiple system atrophy (MSA) and corticobasal degeneration (CBD).





![ Progressive Supranuclear Palsy
Multiple System Atrophy [(Shy-Dragger syn.) SND (MSAP) OPCA (MSAC)]
Corticobasal Degeneration
Dementia with Lewy Body Disease](https://image.slidesharecdn.com/pdplus-180329100935/85/Parkinson-s-plus-syndromes-5-320.jpg)



























































































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































