Inherited myopathies are a diverse group of genetic disorders characterized by primary dysfunction of skeletal muscle. They arise from mutations in genes involved in muscle structure, metabolism, excitation–contraction coupling, or membrane integrity. These conditions are typically present from birth or childhood, though some forms manifest in adolescence or adulthood.
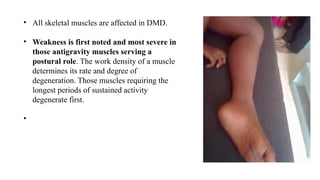
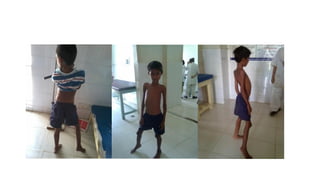
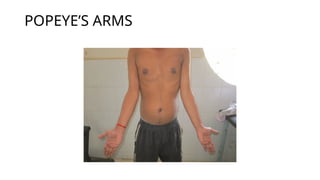
Clinically, inherited myopathies commonly present with muscle weakness, often symmetric and proximal, leading to difficulties with activities such as walking, climbing stairs, or lifting objects. Other features may include muscle wasting, hypotonia, delayed motor milestones, cramps, myalgia, or episodes of exercise intolerance. Certain subtypes are associated with systemic involvement, affecting the heart, respiratory muscles, or eyes.
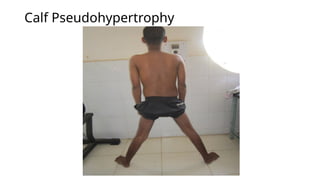
Major categories include muscular dystrophies (e.g., Duchenne and Becker), congenital myopathies (such as central core and nemaline myopathy), metabolic myopathies (including glycogen and lipid storage disorders), and mitochondrial myopathies. Inheritance patterns vary and may be autosomal dominant, autosomal recessive, X-linked, or mitochondrial.
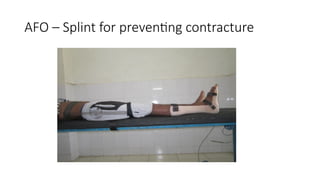
Diagnosis relies on a combination of clinical evaluation, serum creatine kinase levels, electromyography, muscle imaging, muscle biopsy, and increasingly, genetic testing, which has become central to precise classification. While many inherited myopathies have no curative treatment, management focuses on supportive care, physiotherapy, respiratory and cardiac monitoring, and genetic counseling. Advances in molecular medicine are opening avenues for targeted and gene-based therapies.