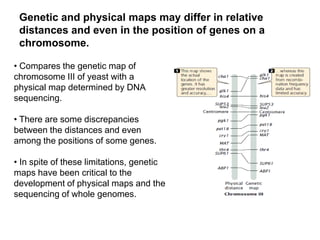
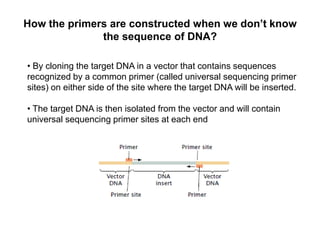
Structural genomics aims to understand genome content through sequencing and mapping genomes. Genetic maps show relative gene locations based on recombination rates, while physical maps use DNA analysis to place genes by base pair distance. Whole genome sequencing involves breaking genomes into fragments that are sequenced and reassembled using overlaps. Functional genomics seeks to identify all genes, RNAs, proteins and their functions through methods like homology searches, microarrays, and mutagenesis screens.