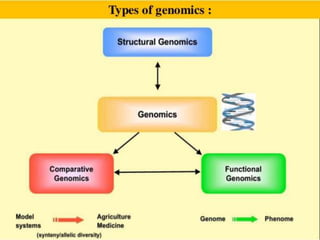
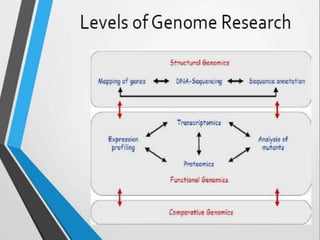
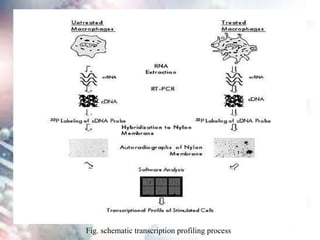
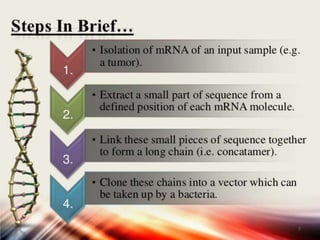
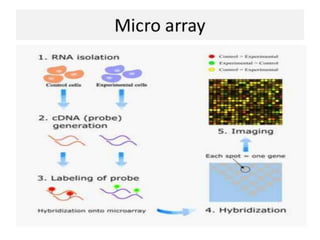
This document discusses functional genomics and its approaches. It defines functional genomics as the worldwide experimental approach to access the function of genes by using information from structural genomics. The key functional genomics approaches discussed are transcriptomics, proteomics, metabolomics, interactomics, epigenetics, and nutrigenomics. Modern techniques discussed include expressed sequence tags (ESTs), serial analysis of gene expression (SAGE), and microarray analysis.





















































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)




