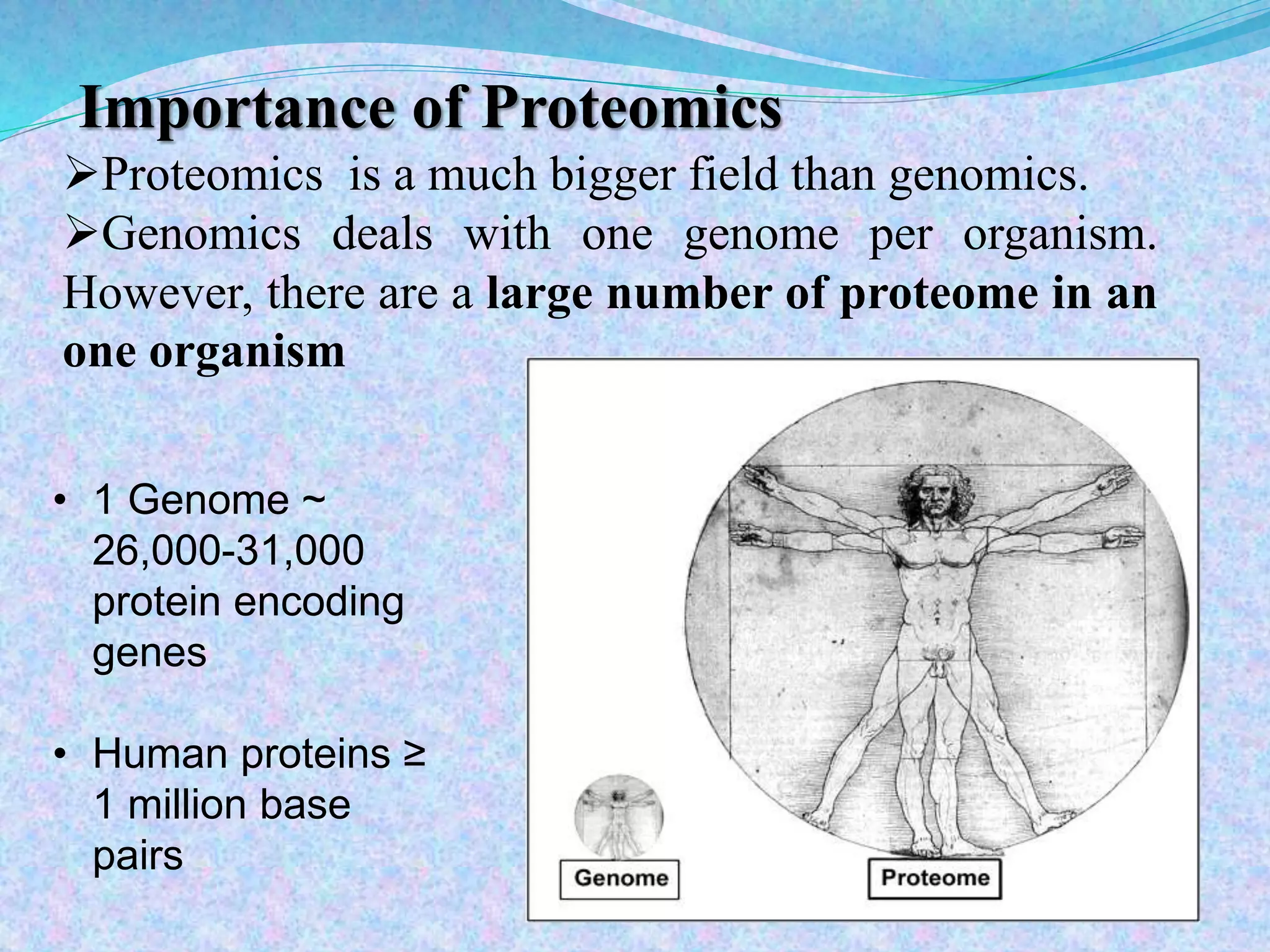
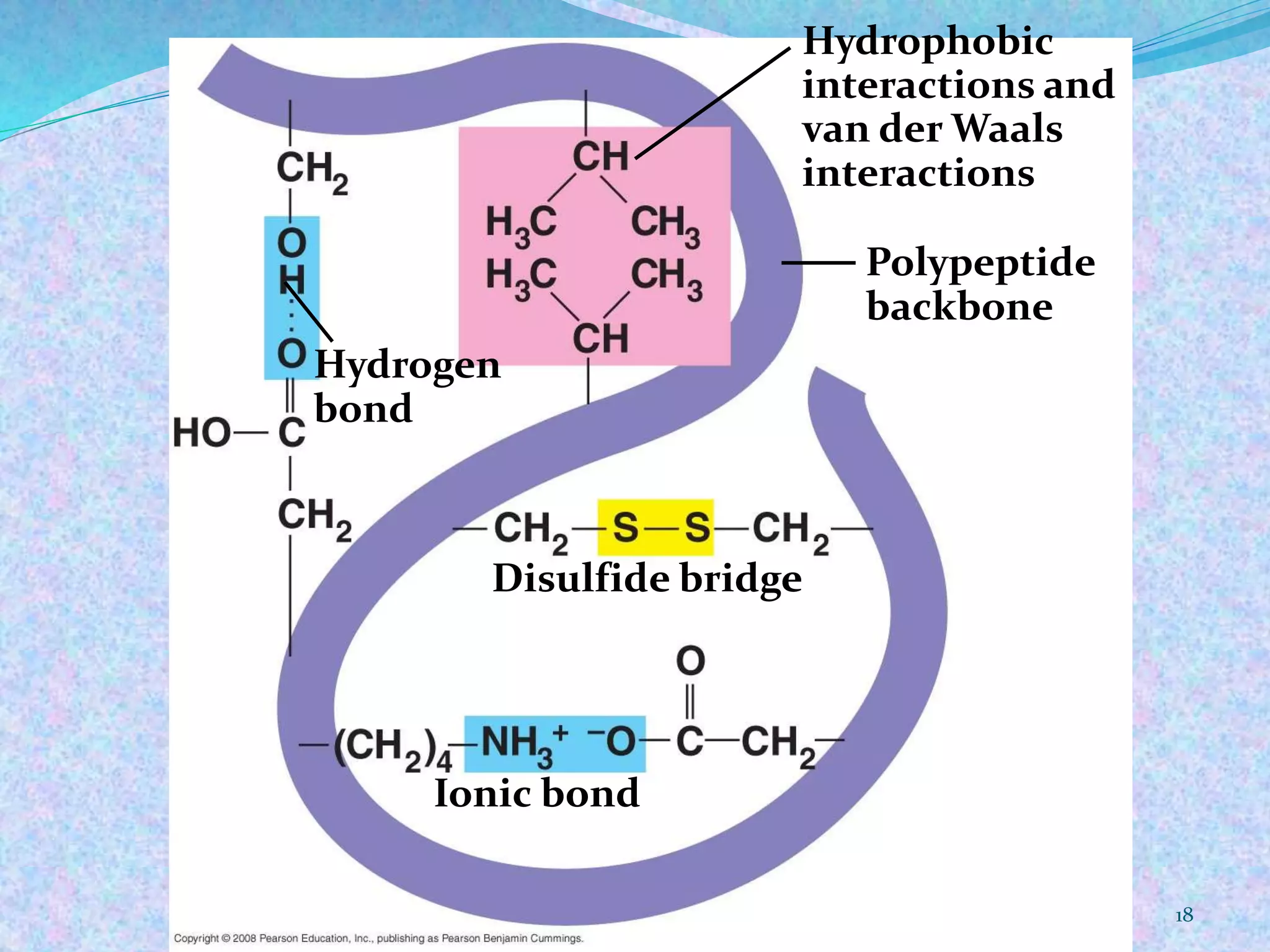
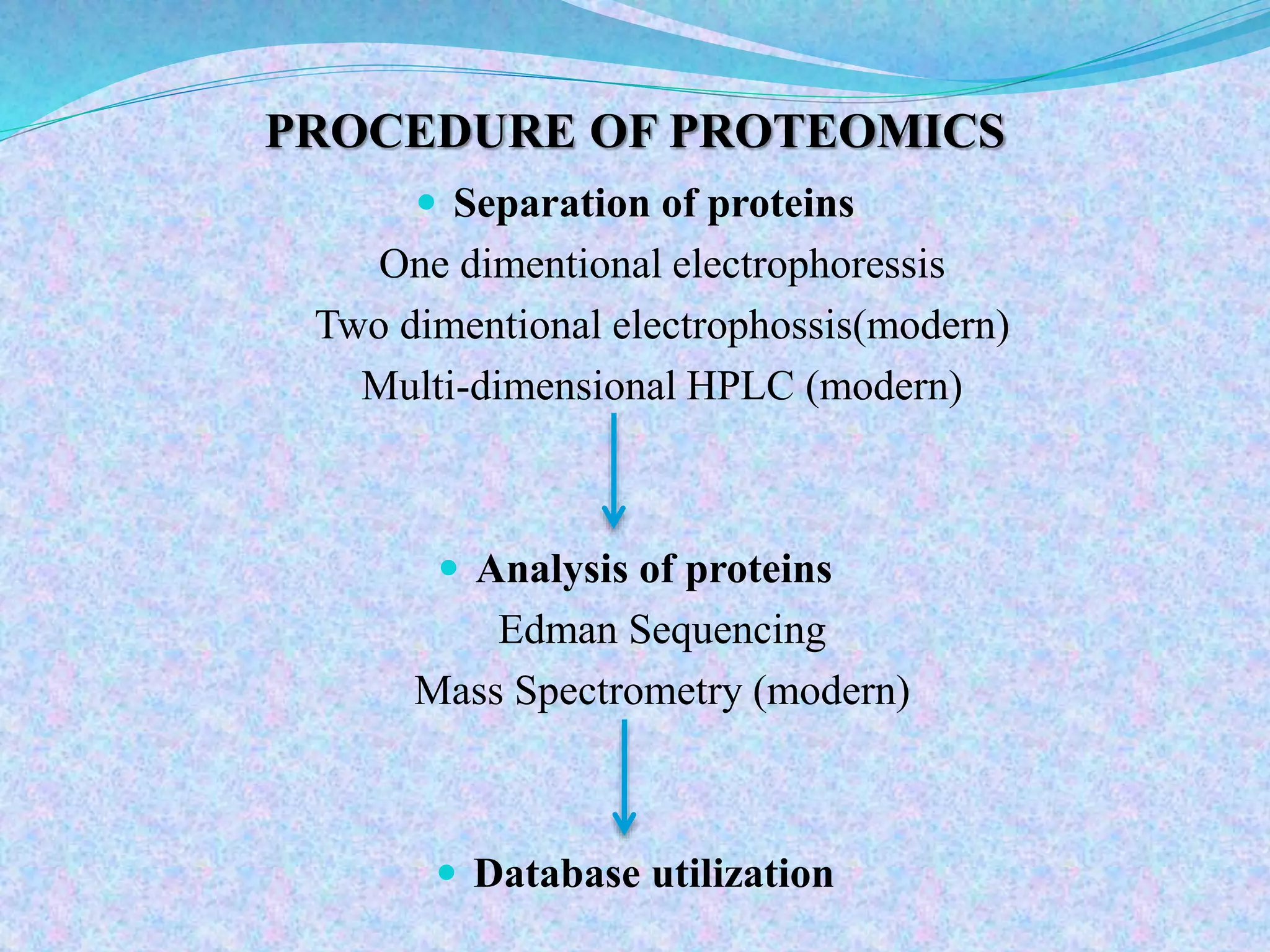
This document provides an overview of proteomics and protein-protein interactions. It begins with an introduction to proteomics, including its history and importance. It then discusses protein structure, including the primary, secondary, tertiary, and quaternary levels. The document outlines different types of proteomics, such as expression, structural, and functional proteomics. It also describes the various steps involved in proteome analysis, including sample preparation, separation, identification, and use of databases. The document discusses techniques for studying protein-protein interactions and provides examples like co-immunoprecipitation and yeast two-hybrid screening. Overall, the document provides a comprehensive overview of the key concepts and methods in the field of proteomics.